


Abstract
Etoposide is a widely prescribed anticancer drug that is, however, associated with an increased risk of secondary leukemia. Although the molecular basis underlying the development of these leukemias remains poorly understood, increasing evidence implicates the interaction of etoposide metabolites [i.e., etoposide quinone (EQ)] with topoisomerase II enzymes. However, effects of etoposide quinone on other cellular targets could also be at play. We investigated whether T-cell protein tyrosine phosphatase (TCPTP), a protein tyrosine phosphatase that plays a key role in normal and malignant hematopoiesis through regulation of Janus kinase/signal transducer and activator of transcription signaling, could be a target of EQ. We report here that EQ is an irreversible inhibitor of TCPTP phosphatase (IC50 = ∼7 μM, second-order rate inhibition constant of ∼810 M−1⋅min−1). No inhibition was observed with the parent drug. The inhibition by EQ was found to be due to the formation of a covalent adduct at the catalytic cysteine residue in the active site of TCPTP. Exposure of human hematopoietic cells (HL60 and Jurkat) to EQ led to inhibition of endogenous TCPTP and concomitant increase in STAT1 tyrosine phosphorylation. Our results suggest that in addition to alteration of topoisomerase II functions, EQ could also contribute to etoposide-dependent leukemogenesis through impairment of key hematopoietic signaling enzymes, such as TCPTP.
Introduction
Protein tyrosine phosphatases (PTPs) are important regulators of the activity of numerous signaling pathways involved in major cellular processes such as cell growth, proliferation, and differentiation (Tonks, 2006; Tiganis and Bennett, 2007; Pike and Tremblay, 2016). T-cell protein tyrosine phosphatase (TCPTP) is a cytosolic tyrosine phosphatase which is ubiquitously expressed. However, the highest levels of expression of TCPTP are found in hematopoietic cells, where this enzyme modulates growth factor and cytokine signaling pathways, thus contributing to immune and hematopoietic cell homeostasis (Wiede et al., 2012; Pike and Tremblay, 2016). In particular, TCPTP negatively regulates Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through the dephosphorylation of different tyrosine phosphorylated JAK/STAT proteins, such as STAT1 or JAK1 (ten Hoeve et al., 2002; Dorritie et al., 2014; Pike and Tremblay, 2016). In addition to STAT1, TCPTP also dephosphorylates STAT3 and STAT5 and negatively regulates their activation (Pike and Tremblay, 2016). The JAK/STAT pathway plays a crucial role in hematopoiesis, and aberrant activation of STAT signaling is involved in leukemogenesis (Dorritie et al., 2014; Pike and Tremblay, 2016). Interestingly, deletions or inactivating mutations of TCPTP were identified in T-cell leukemia and non-Hodgkin’s lymphoma and associated with elevated STAT signaling and changes in gene expression (Kleppe et al., 2010, 2011a,b; Pike and Tremblay, 2016). In addition, it has been reported that TCPTP is overexpressed in MYC-driven mouse B-cell lymphoma (Young et al., 2009). The importance of TCPTP in hematopoiesis is further supported by knockout mouse models (PTPN2−/−), where the absence of TCPTP induces severe hematopoietic defects (affecting lymphoid, myeloid, and erythroid lineages) and progressive systemic inflammation leading to death (You-Ten et al., 1997; Bourdeau et al., 2007; Heinonen et al., 2009; Wiede et al., 2012). These studies highlight the important role of TCPTP in normal and malignant hematopoiesis.
Etoposide (ETOP) is a widely used anticancer drug to treat a variety of human malignancies (Hande, 1998; Baldwin and Osheroff, 2005). The mechanisms of action proposed for its antitumor activity are based mainly on its interaction with topoisomerase II (Baldwin and Osheroff, 2005; Deweese and Osheroff, 2009). ETOP is indeed known to affect the catalytic cycle of these enzymes and to stabilize topoisomerase II–bound DNA strand breaks, which have the potential to activate cell death pathways (Jacob et al., 2011). Despite the wide clinical use of ETOP, this chemotherapeutic drug is known to induce treatment-related leukemias (Baldwin and Osheroff, 2005; Pendleton et al., 2014). In humans, ETOP can be oxidized by cytochrome P450 (CYP3A4) and myeloperoxidases into etoposide quinone (EQ), which was found to have an effect on topoisomerase II enzymes several times stronger than that of the parent drug (Zhuo et al., 2004; Fan et al., 2006; Jacob et al., 2011; Vlasova et al., 2011). Although the molecular basis for ETOP-induced leukemogenesis is not well understood, evidence increasingly indicates that EQ is a critical contributor to the development of ETOP-related secondary leukemia, in particular through alteration of topoisomerase II functions (Gantchev and Hunting, 1998; Kagan et al., 1999; Fan et al., 2006; Vlasova et al., 2011; Smith et al., 2014; Gibson et al., 2016). However, interactions of ETOP reactive metabolites with other cellular proteins and macromolecules may also contribute to ETOP-dependent leukemogenesis (Fan et al., 2006; Rojas et al., 2009).
We show here that EQ is an irreversible inhibitor of TCPTP phosphatase activity. Kinetic and molecular analyses using purified human TCPTP indicated that the irreversible inhibition of the enzyme by EQ is due to the formation of a covalent adduct with its catalytic cysteine. Accordingly, exposure of cultured hematopoietic cells to EQ leads to the irreversible inhibition of the endogenous TCPTP with a concomitant increase in cellular STAT1 phosphorylation. Interestingly, it is well known that dysregulation of the JAK/STAT signaling pathway is involved in leukemogenic processes (Benekli et al., 2009; Dorritie et al., 2014). Altogether, our data suggest that in addition to the disruption of topoisomerase II functions, EQ may also contribute to ETOP-related leukemia through alteration of important hematopoietic signaling enzymes, such as TCPTP.
Materials and Methods
Chemicals and Cells.
ETOP and EQ were obtained from Toronto Research Chemicals (North York, Canada). Hydrogen peroxide, N-ethylmaleimide, fluorescein 5-iodoacetamide (5-IAF), myeloperoxidase (MPO), p-nitrophenyl phosphate (pNPP), sodium orthovanadate, nitroblue tetrazolium (NBT), and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (France). ETOP and EQ were diluted in DMSO at a stock concentration of 100 mM. HL60 (human acute promyelocytic leukemia) and Jurkat (human acute T-cell leukemia) cells were from Sigma-Aldrich.
Expression and Purification of Recombinant Human TCPTP Enzyme.
Human TCPTP enzyme was expressed and purified as previously described (Duval et al., 2015), using BL21 (DE3) Escherichia coli, and transformed with a pET28a plasmid containing the cDNA of human PTNP2 (TC45 variant). The purified enzyme was reduced by incubation with 10 mM dithiothreitol (DTT) for 10 minutes in ice before buffer exchange with a PD-10 column (GE Healthcare, France) into 25 mM Tris-HCl, 150 mM NaCl, pH 7.5 (reaction buffer). The protein concentration was determined by the Bradford reagent (Bio-Rad, France), and purity was assessed by SDS-PAGE and Coomassie staining. Purified recombinant human TCPTP (1 mg/ml) was stored at −80°C until use.
Determination of TCPTP Activity Using pNPP Assay.
The phosphatase activity was measured using pNPP as substrate as described previously (Montalibet et al., 2005). Typically, samples containing TCPTP were diluted 10 times with 100 mM sodium acetate buffer (pH 6, 1 mM DTT) containing 5 mM pNPP. The formation of the product (p-nitrophenol) was tracked by continuous measurement of the absorbance at 405 nm at 37°C using a thermostatic microplate reader (BioTek, France) in a total volume of 250 μl. The final concentration of TCPTP during the assay was 100 nM.
Determination of TCPTP Activity Using a Tyrosine-Phosphorylated STAT1 Peptide (Fluorescein Amadite-Phosphorylated STAT1).
The tyrosine phosphatase activity of TCPTP was measured by reverse-phase ultrafast liquid chromatography (RP-UFLC) using a fluorescein amadite (FAM)-conjugated peptide derived from the sequence of human STAT1 (697KGTGYIKTE705, where the Y701 is phosphorylated) as described previously (Duval et al., 2015). In brief, samples containing TCPTP were diluted 100-fold with acetate buffer. Aliquots (100 μl) were incubated for 30 minutes at 37°C in the presence of 50 μM FAM–phosphorylated STAT1 peptide (final concentration of TCPTP was 10 nM). The reaction was stopped with 100 μl of 15% (v/v) HClO4 prior to analysis by RP-UFLC using a Shim-pack XR ODS column (Shimadzu, France) connected to a Prominence Shimadzu UFLC system.
Effects of Etoposide and Etoposide Quinone on TCPTP Activity.
Recombinant TCPTP (1 µM) was incubated with ETOP or different concentrations of EQ in 100 mM sodium acetate, pH 6, for 30 minutes at 37°C (total volume of 50 μl). Samples were diluted 10 times, then assayed for residual TCPTP activity using pNPP. To obtain the IC50 value, the dose-response curves were fitted with the Hill equation, TCPTPactivity = 100/(1 + 10^((logIC50 − [EQ]) × Hillslope)), where IC50 is the half-maximal inhibitory concentration of EQ. The effects of ETOP and EQ on the TCPTP tyrosine phosphatase activity were also assessed by an RP-UFLC approach using a tyrosine phosphorylated STAT1 peptide (see previous section).
Effects of Reducing Agents and Buffer Exchange on TCPTP Inhibited by EQ.
TCPTP (1 µM) was first incubated with EQ (40 µM) or ETOP (100 μM) in 100 mM sodium acetate, pH 6, for 30 minutes at 37°C (total volume of 50 μl). Samples were either incubated with 1 or 10 mM DTT (10 minutes at room temperature) or buffer exchanged (PD Spin Trap G-25; GE Healthcare) to 100 mM sodium acetate, pH 6, prior to measurement of residual TCPTP activity using pNPP.
Fluorescein-Conjugated Iodoacetamide Labeling, Nitroblue Tetrazolium Staining, and Detection of Oxidized TCPTP Catalytic Cysteine.
TCPTP (1 µM) was first incubated with ETOP (100 µM) or EQ (40 µM) in 100 mM sodium acetate, pH 6, at 37°C for 30 minutes followed by the addition of 20 μM 5-IAF (total volume of 50 μl) and further incubation at 37°C for 10 minutes (in the dark). Samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. 5-IAF covalent labeling of TCPTP cysteine residues was detected by western blot using antifluorescein antibodies (reference number 11426346910; Sigma-Aldrich). Membranes were stripped and further probed with anti-TCPTP antibody (reference number SAB4200249; Sigma-Aldrich).
The formation of covalent adducts of EQ on TCPTP was detected by NBT redox-cycling staining as reported previously (Paz et al., 1991). In brief, TCPTP (1 µM) was incubated with ETOP (100 µM) or EQ (40 µM) in 100 mM sodium acetate, pH 6, at 37°C for 30 minutes (total volume 50 μl). Samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. EQ-bound TCPTP on the membranes was stained by incubation with 0.6 mg/ml NBT in 2 M potassium glycinate, pH 10, for 30–45 minutes in the dark at room temperature. Next, membranes were washed thoroughly with PBS and further probed with anti-TCPTP antibody (reference number 11426346910; Sigma-Aldrich).
For the detection of oxidized TCPTP catalytic cysteine, TCPTP (1 μM) was treated with ETOP (100 μM), EQ (40 μM), or H2O2 (100 μM) in a total volume of 50 μl. Aliquots were then separated by SDS-PAGE and analyzed by western blot with a specific antioxidized PTP active site antibody (reference number MAB 2844; R&D Systems) as described previously (Duval et al., 2015).
Effects of MPO-Dependent Bioactivation of ETOP into EQ.
Myeloperoxidase was used to bioactivate ETOP into EQ as described previously (Kagan et al., 1999; Fan et al., 2006; Vlasova et al., 2011). To this end, MPO (5 U) was incubated with ETOP (100 µM) and H2O2 (100 µM) in 100 mM sodium acetate, pH 6.5, for 30 minutes at room temperature (in a total volume of 1 ml). Heat-inactivated MPO (boiled for 30 minutes) was used as a negative control. Catalase (300 U/ml final concentration) was added to remove excess H2O2. Finally, reaction mix (48 μl) was added to recombinant TCPTP (1 µM final concentration) in 100 mM sodium acetate, pH 6, and incubated for 30 minutes at 37°C (total volume of 50 μl). After incubation, and a 10-times dilution, the residual activity of TCPTP was measured using the pNPP assay. IAF and NBT labeling and detection of oxidized TCPTP catalytic cysteine were also carried out on TCPTP treated with MPO as described earlier.
Kinetics of TCPTP Inhibition by EQ.
The kinetic data were analyzed as reported by Copeland (2005) for irreversible inhibitors. The kinetic data were fitted and plotted using Qtiplot software (http://www.qtiplot.com/). In brief, TCPTP (3 µM) was incubated with different concentrations of EQ (pseudo first-order conditions) in 100 mM sodium acetate, pH 6, at 37°C. At different time points, aliquots were removed and assayed for residual TCPTP activity using pNPP assay. The rate of inhibition of TCPTP by EQ can be represented as follows: ln [% residual activity] = kobs × t (where t is time and kobs is the apparent first-order inhibition rate constant). The apparent first-order inhibition rate constants (kobs = kinact × [EQ]) can be calculated for each EQ concentration from the slope of the natural log (ln) of percentage residual activity plotted against time. The second-order rate constant (kinact) was determined from the slope of kobs plotted against EQ concentrations.
Effects of EQ on TCPTP in the Presence of the Competitive PTP Inhibitor Orthovanadate (Na3VO4).
TCPTP (1 µM) was incubated with 1 mM orthovanadate in the presence or absence of EQ (40 µM) in 100 mM sodium acetate, pH 6, at 37°C for 30 minutes. Samples were diluted 10 times in 100 mM sodium acetate, pH 6, containing 1 mM EDTA and residual TCPTP activity measured using pNPP assay.
Molecular Docking.
TCPTP protein structure data were obtained from the RCSB Protein Data Bank database (Protein Data Bank ID 1L8K). The protein structure was prepared using the PrepWizard module in the Schrödinger suite (Schrödinger LLC, https://www.schrodinger.com). In brief, missing side chains or loops were completed. The pKa values of residues were predicted and hydrogens were added at pH 7. Water molecules were removed. Finally, the structure was optimized by a restrained energy minimization with the OPLS3 force field. The ligand (EQ) was constructed by a Maestro module and was also refined by the OPLS3 force field. Covalent docking was carried out with the catalytic residue Cys 216 selected as the docking center. Grid box size was set to 20 × 20 × 20 Å3. Michael addition reaction type was selected for the covalent docking. The final conformation was selected, and images were prepared with VMD program (Humphrey et al., 1996).
Mass Spectrometry Analysis.
TCPTP (1 µM) was incubated with 100 µM EQ for 30 minutes at 37°C in 100 mM sodium acetate, pH 6. After reduction with DTT (10 mM), the samples were diluted 10 times in sodium acetate buffer, and unmodified thiol moieties of cysteines were blocked by addition of 10 mM N-ethylmaleimide for 10 minutes. Samples were then incubated overnight at 37°C with trypsin (Promega, France) at 12.5 ng/μl in 25 mM ammonium bicarbonate, pH 8.0. The supernatant containing peptides was acidified with formic acid (FA); desalted on C18 tips (Pierce C18 tips; Thermo Scientific, France); and eluted in 10 µl of 70% acetonitrile, 0.1% FA. Desalted samples were evaporated using a SpeedVac, then taken up in 10 µl of buffer A (buffer A: water, 0.1% FA), and 5 µl were injected on a nanoLC high-performance liquid chromatography system (Thermo Scientific) coupled to a hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific). Peptides were loaded on a reverse-phase C18 µ-precolumn (C18 PepMap100, 5 µm, 100 A, 300 µm i.d. × 5 mm) (ThermoFischer, Paris France) and separated on a C18 column (EASY-spray C18 column, 75 µm i.d. × 50 cm) (ThermoFischer) at a constant flow rate of 300 nl/min, with a 120-minute gradient of 2%–40% buffer B (buffer B: 20% water, 80% acetonitrile, 0.1% FA). Mass spectrometry (MS) analyses were performed by the Orbitrap cell with a resolution of 120,000 (at m/z 200). Tandem MS (MS/MS) fragments were obtained by higher-energy collisional dissociation activation (collisional energy of 28%) and acquired in the ion trap in top-speed mode with a total cycle of 3 seconds. The database search was performed against the Swiss-Prot database (02/2017) and the Homo sapiens taxonomy with Mascot v2.5.1 software (Matrix Sciences, Boston, MA) with the following parameters: tryptic peptides only with up to two missed cleavages, variable modifications: cysteine EQ and methionine oxidation. MS and MS/MS error tolerances were set to 7 ppm and 0.5 Da, respectively. Peptide identifications were validated using a 1% false discovery rate threshold obtained by Proteome Discoverer (version 2.2; Thermo Scientific) and the percolator algorithm. The candidate sequences modified by EQ were manually inspected for de novo sequencing.
Cell Culture, TCPTP Immunoprecipitation, and STAT1 Phosphorylation Kinetics.
HL60 and Jurkat cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum and 1 mM l-glutamine in T-75 flasks. Cells (60 ml at 2 × 106 cells/ml) were washed with PBS prior to exposure to 50 μM EQ (or DMSO) for 30 minutes at 37°C (5% CO2) in RPMI 1640 medium.
For immunoprecipitation of endogenous TCPTP, treated cells were washed with PBS and resuspended in lysis buffer (PBS, 1% Triton X-100, 1 mM sodium orthovanadate, phosphatase cocktail inhibitor 2, and protease inhibitor cocktail; Sigma-Aldrich) for 20 minutes. Cell lysates were centrifuged at 15,000g for 10 minutes at 4°C, and supernatants (whole cell extracts) were taken. TCPTP was immunoprecipitated by incubating 1 mg of whole cell extracts with 1 µg of polyclonal TCPTP antibody (reference number SAB4200249; Sigma-Aldrich) overnight at 4°C. Samples were then rocked for 1 hour at 4°C in the presence of 30 μl of protein A–Agarose (Santa Cruz, Germany). The immunobeads were harvested by centrifugation, washed three times with lysis buffer, and split into two portions. One part of the beads was incubated with 50 µM FAM–phosphorylated STAT1 peptide to measure residual immunoprecipitated-TCPTP activity by RP-UFLC as described earlier. The second part of immunobeads was incubated with nonreducing Laemmli sample buffer. Eluates were separated by SDS-PAGE and analyzed by western blot using an anti-TCPTP antibody (reference number MAB 1930; R&D Systems).
For the STAT1 phosphorylation kinetic analysis, cells treated with EQ or DMSO were washed with fresh medium and incubated for 20 minutes in medium supplemented with 10 ng/ml of human interferon γ (IFNγ). Cells were then washed in fresh medium, and aliquots (2 × 106 cells) were removed at different time points, washed with PBS, and lysed. STAT1 activation was then assayed in lysates by western blot using anti-phospho-STAT1 (phosphorylated tyrosine 701, reference number 9176; Cell Signaling Technology). Membranes were reprobed sequentially with anti-STAT1 (reference number 9167; Cell Signaling Technology), anti-TCPTP (reference number SAB4200249; Sigma-Aldrich), and anti-β actin (reference number 3700; Cell Signaling Technology) antibodies. Quantification of STAT1 phosphorylation in western blots was carried out using ImageJ software (National Institutes of Health) (Schneider et al., 2012).
Statistical Analysis.
The study was designed in such a manner that three independent replicates were conducted for each experiment prior to statistical analysis. All data are expressed as the mean ± S.D. All statistical analyses were then carried out using Prism 5.03 (GraphPad Software, La Jolla, CA) with a threshold of P < 0.05 to consider differences as statistically significant. For experiments where multiple groups were compared with control, a one-way analysis of variance (ANOVA) was used. If statistical significance was reached, data were further analyzed with Dunnett’s post hoc test. For experiments between two groups, a two-tailed Student’s t test was used. No experiment was removed from the final analysis. For quantitative polymerase chain reaction experiments (Supplemental Fig. 3), six experimental points were used in the data analysis, which consisted of a Student’s t test to compare the effect of EQ versus control in cells induced with interferon γ.
Results
Effect of Etoposide Quinone on Activity of TCPTP.
The effects of ETOP and EQ on the activity of recombinant human TCPTP expressed and purified from bacteria were assessed. As a first experiment, TCPTP was incubated in the presence of 100 µM ETOP or increasing concentrations of EQ prior to the measurement of the residual enzyme activity with the chromogenic pNPP phosphatase substrate. As shown in Fig. 1A, EQ inhibits the hydrolysis of this substrate in a dose-dependent manner with half-maximal inhibitory concentration (IC50) values of 7.3 μM (Supplemental Fig. 1). Conversely, 100 µM ETOP did not display any inhibitory effect, therefore suggesting that inhibition of TCPTP activity could occur with EQ but not with the parent drug. These results were further validated with a UFLC-based enzyme assay using a more specific TCPTP substrate consisting of a phospho-STAT1 peptide derived from the sequence of human STAT1 (697KGTGYIKTE705, where the Y701 is phosphorylated) as described by Duval et al. (2015). UFLC quantification of the dephosphorylated STAT1 confirms that EQ, but not ETOP, inhibits TCPTP phosphatase activity (Fig. 1B).

Effect of ETOP and EQ on human TCPTP activity. (A) Effect of ETOP and EQ on TCPTP activity measured by pNPP dephosphorylation. One micromolar TCPTP was incubated with 100 µM ETOP and 5–40 µM EQ for 30 minutes at 37°C and diluted 10-fold prior to measurement of residual activity in the presence of 5 mM pNPP as described in the Materials and Methods section. Results are the mean of three independent experiments; error bars indicate S.D. (B) Measurements of TCPTP FAM–phosphorylated STAT1 peptide dephosphorylation activity. TCPTP was incubated with 100 µM ETOP and 40 µM EQ for 30 minutes at 37°C and residual TCPTP activity measured as described under Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D. *P < 0.05, determined using ANOVA followed by Dunnett’s post hoc analysis. Ctrl, control.
Etoposide Quinone Covalently Reacts with TCPTP Cysteine Residues.
As EQ, but not ETOP, inhibits TCPTP activity, we next hypothesized that this inhibition could result from the general chemical reactivity properties of quinone chemicals and, notably, the possibility that they react covalently with proteins (Bolton and Dunlap, 2017; Bolton et al., 2018). NBT can be used to stain proteins covalently modified by quinones to form quinone proteins. As shown in Fig. 2A, NBT reaction generated only a signal when TCPTP was treated with EQ, confirming the presence of covalent quinone adducts on TCPTP.

Modification of TCPTP cysteine residues by EQ. (A) Detection of human TCPTP adducts by NBT staining. Human TCPTP was incubated with 40 µM EQ or 100 µM ETOP for 30 minutes. Samples were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Quinone adducts were detected by NBT staining as described in Materials and Methods. The membrane is representative of three independent experiments. (B) 5-IAF staining of human TCPTP to detect unmodified cysteines. Human TCPTP was incubated with 40 µM EQ or 100 µM ETOP for 30 minutes prior incubation for 10 minutes with 20 μM 5-IAF. SDS-PAGE was transferred onto a nitrocellulose membrane. IAF adducts were detected by fluorescence after SDS-PAGE as described in the Materials and Methods section. The membrane is representative of three independent experiments. (C) Detection of TCPTP protein oxidation. Human TCPTP was incubated with 100 µM ETOP, 40 µM EQ, or 100 µM H2O2 for 30 minutes prior to analysis with western blot using an antioxidized TCPTP active site antibody as described in Materials and Methods. The membrane is representative of three independent experiments. Ctrl, control.
Knowing that protein cysteinyl residues are common nucleophilic targets of quinone electrophiles, we therefore strived to show whether TCPTP cysteinyl residues could be modified after exposure to EQ. TCPTP was treated with EQ and then incubated with fluorescein-conjugated iodoacetamide (IAF), a specific cysteinyl thiol reagent. Our results (Fig. 2B) show that EQ decreased IAF labeling of TCPTP, therefore indicating that EQ reacts with cysteine residues of TCPTP.
Quinone chemicals can also display redox-cycling properties that lead to generation of reactive oxygen species. In addition, it is known that TCPTP can be inhibited by reactive oxygen species through the oxidation of its catalytic cysteine residue (Ostman et al., 2011). We therefore evaluated whether EQ could oxidize the catalytic cysteine residue of TCPTP using an antibody specifically directed against the oxidized catalytic cysteine residue of protein tyrosine phosphatases (Duval et al., 2015). Figure 2C shows that EQ treatment does not lead to TCPTP active site cysteine oxidation, and that the basal oxidation of catalytic cysteine achieved during the experiment and observed in control and ETOP treatment is decreased with EQ. This is in agreement with the formation of an EQ adduct on the catalytic cysteine of the enzyme. Therefore, these data altogether suggest a loss of TCPTP activity due to EQ adduction of the catalytic cysteine.
When clinically administered to patients, ETOP is biochemically metabolized to EQ by different enzymes, notably myeloperoxidase, which is highly expressed in bone marrow hematopoietic cells (Fan et al., 2006; Atwal et al., 2017). To test if EQ generated from peroxidase/H2O2-dependent bioactivation of ETOP would yield similar TCPTP inhibition, we used an in vitro peroxidase activation system that mimics the bioactivation of ETOP into EQ in hematopoietic cells described previously (Kagan et al., 1999; Vlasova et al., 2011). As shown in Supplemental Fig. 2A, in the presence of a functional peroxidase/H2O2 enzymatic system, ETOP is capable of inhibiting TCPTP activity with concomitant quinone adduction (NBT labeling) and loss of IAF cysteine labeling of TCPTP protein (Supplemental Fig. 2, B and C). Conversely, when inactive peroxidase is used, no inhibition of TCPTP and quinone adduction on cysteine residues is observed (Supplemental Fig. 2). This confirms that EQ, but not the parent drug, can inhibit TCPTP activity through covalent adduction, a result which is consistent with the electrophilic nature of the quinone moiety of EQ which enables Michael addition with thiol groups (Fan et al., 2006; Bolton et al., 2018).
Identification of the Catalytic Cysteine Residue of TCPTP as a Target of EQ.
Orthovanadate, a transition state analog generally used as a competitive protein phosphotyrosine phosphatase inhibitor, was used to evaluate whether EQ inhibition involves reactions at the active site of the enzyme as described previously (Seiner et al., 2007). Figure 3A shows that vanadate confers protection to TCPTP as the inhibition of the enzyme is significantly slowed by the competitive inhibitor, thereby providing evidence that the reaction is active site directed. Computational analysis using covalent docking approaches further indicated that the catalytic cysteine of TCPTP (cysteine 216) can form an adduct in the active site through the formation of a covalent bond between the sulfur atom and the carbon 6′ of the quinone moiety (Fig. 3B).

Mapping of EQ adduct on TCPTP protein active site cysteine. (A) Effect of sodium orthovanadate on TCPTP inhibition by EQ. One micromolar human recombinant TCPTP was incubated with 40 µM EQ and/or 1 mM orthovanadate for 30 minutes at 37°C and residual activity measured as described in Materials and Methods. The experiment is representative of three independent experiments. (B) Molecular docking model of EQ bound in a human TCPTP protein structure. EQ molecule adduct atom coordinates were obtained inside the TCPTP active site pocket as described in the Materials and Methods section. EQ is displayed with green sticks and is covalently bonded to the C216 residue. (C) Mass spectrometry characterization of EQ adduct on the active site cysteine tryptic peptide. One micromolar TCPTP protein was incubated with 100 µM EQ for 30 minutes at 37°C prior to trypsine treatment and liquid chromatography–MS/MS analysis as described in Materials and Methods. The upper panel depicts the spectrum obtained with control, untreated TCPTP protein, whereas the lower panel shows data acquired with TCPTP samples treated with EQ. The sequence of the peptide is displayed on each panel in addition to the position of the EQ adduct on cysteine in the lower panel. Abs, absorbance; AU, arbitrary units.
To ascertain that the catalytic cysteine residue of TCPTP can be covalently adducted by EQ, we analyzed recombinant protein treated with EQ using liquid chromatography–MS/MS. As shown in Fig. 3C, the [M+3H]3+ molecular ion of the tryptic peptide harboring the catalytic cysteine displays a 190.9 m/z increase after treatment with EQ, corresponding to the mass of the EQ adduct. This mass of EQ adduct is also confirmed in the a162+, y172+, and y203+ ions of the fragmented peptide. It is well known that tyrosine phosphatases are readily inhibited by chemicals able to modify their catalytic cysteine (Seiner et al., 2007; Ostman et al., 2011). Altogether, these experiments indicate that EQ impairs TCPTP activity through the covalent adduction of its catalytic cysteine.
Characterization of Reversibility and Kinetics of TCPTP Inhibition by EQ.
TCPTP inhibition by EQ was further evaluated for reversibility. To this end, TCPTP enzyme was inhibited by EQ and subsequently subjected to a buffer exchange experiment before measuring its residual activity. Consistent with the data described earlier, buffer exchange could not restore TCPTP activity (Fig. 4A). Depending on the chemical structure of the quinones, reversibility of cysteine adducts through reduction has already been described (Bolton and Dunlap, 2017; Bolton et al., 2018). We therefore examined the effect of two concentrations of the reducing agent DTT on the reversion of TCPTP inhibition by EQ. As shown in Fig. 4B, even at a 10 mM concentration, DTT was not successful at restoring the activity of EQ-inhibited TCPTP, thereby confirming the stable and covalent nature of the reaction between EQ and the catalytic cysteine of TCPTP. These observations are consistent with previously published data on the inhibition of topoisomerase II by EQ (Jacob et al., 2011; Smith et al., 2014; Gibson et al., 2016).

Irreversibility of TCPTP inhibition by EQ. (A) Effect of buffer exchange on activity of TCPTP preincubated with EQ. Human TCPTP was incubated with 40 µM EQ or 100 µM ETOP for 30 minutes followed by buffer exchange or no buffer exchange prior to residual pNPP dephosphorylation activity determination as described in Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D. *P < 0.05, determined using ANOVA followed by Dunnett’s post hoc analysis. (B) Effect of DTT on activity of TCPTP preincubated with EQ. Human TCPTP was preincubated or not with 40 µM EQ for 30 minutes followed by treatment with 1 or 10 mM DTT. Residual pNPP dephosphorylation activity was determined as described in Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D. *P < 0.05, determined using ANOVA followed by Dunnett’s post hoc analysis. Ctrl, control.
Finally, the kinetics of TCPTP inhibition by EQ were studied under pseudo first-order conditions, which allowed observation of time- and dose-dependent inhibition (Fig. 5A) and permitted the determination of the second-order inhibition kinetic constant, kinact = ∼810 ± 73 M−1⋅min−1, derived from the slope of the graph with pseudo first-order kinetic constants plotted against EQ concentration (Fig. 5B). These kinetic data also confirm the irreversible nature of the inhibition of TCPTP by EQ.

Kinetics of TCPTP inhibition by EQ. (A) Determination of pseudo first-order inhibition rate constant for TCPTP inhibition by EQ. TCPTP (3 µM) was incubated with different concentrations (0, 10, 20, and 40 µM) of EQ and assayed for residual activity at different time points as described in Materials and Methods. The value of kobs was determined as the slope of the linear regression of data expressed as the natural logarithm of the residual activity as a function of time. Results are the mean of three independent experiments; error bars indicate S.D. (B) Determination of the second-order inhibition rate constant for TCPTP inhibition by EQ. Values of kobs were plotted as a function of EQ concentration, and kinact was determined as the slope of the linear regression as described in Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D.
EQ Alters TCPTP Activity and Enhances STAT1 Phosphorylation in HL60 and Jurkat Cells.
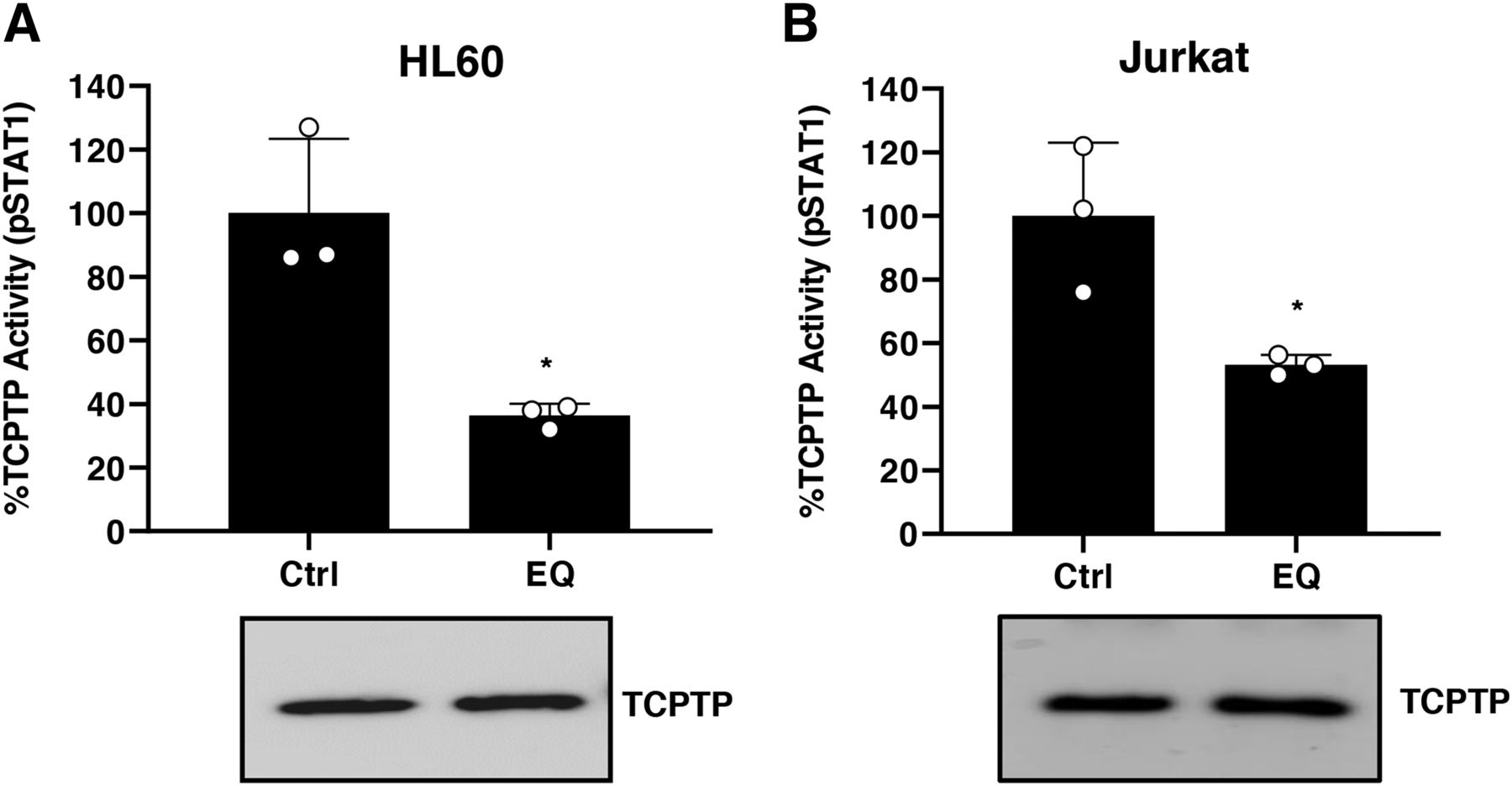
Taking into consideration the in vitro effects of EQ observed with recombinant TCPTP protein, HL60 and Jurkat cell lines were treated with EQ to study its effect on cellular TCPTP. After 30-minute exposure, the cells were lysed and cellular TCPTP immunoprecipitated. The residual TCPTP activity was measured using the phospho-STAT1 peptide substrate dephosphorylation assay using UFLC detection (Duval et al., 2015) and immunoprecipitated-TCPTP protein content evaluated by western blot. Figure 6 shows that, upon exposure to EQ, the cellular TCPTP activity decreased to roughly 30% and 50% that of the control cells in HL60 and Jurkat cells, respectively. These data are consistent with studies carried out on the inhibition of PTP1B (a tyrosine phosphatase related to TCPTP) by naphthoquinones (Iwamoto et al., 2007). These results indicate that EQ inhibits TCPTP in a cellular environment, and we therefore investigated whether, under such conditions, TCPTP-dependent intracellular signaling could be altered. Incubation of HL60 cells with IFNγ triggers STAT1 phosphorylation signaling. This signaling is sustained at a higher level (1.5-fold for 60 minutes) in cells exposed to EQ treatment as compared with untreated controls (Fig. 7A). Similar results were also obtained with Jurkat cells, with more than 2-fold phosphorylation after 1 hour (Fig. 7B). To further show that EQ increases STAT1 signaling, we measured the expression of three genes (APOL1, GBP1, and IRF1) known to be regulated by STAT1 (Hartman et al., 2005; Reardon and McKay, 2007). As shown in Supplemental Fig. 3, quantitative polymerase chain reaction analyses indicated that the expression of APOL1, GBP1, and IRF1 is increased in Jurkat cells exposed to EQ. These results indicate that inhibition of TCPTP by EQ is associated with increased STAT1 phosphorylation and higher expression of STAT1-regulated genes in these cells upon stimulation with IFNγ.

Inhibition of TCPTP by EQ in HL60 and Jurkat cells. HL60 (A) and Jurkat cells (B) were treated or not [control (Ctrl)] with 50 µM EQ for 30 minutes at 37°C. Cells were lysed and cellular TCPTP protein was immunoprecipitated prior to residual activity determination and TCPTP protein content determination by western blotting as described in Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D. *P < 0.05, determined using Student’s t test as described under the Materials and Methods section. Western blot is representative of three independent experiments. pSTAT1, phosphorylated STAT1.

STAT1 phosphorylation kinetic analysis in HL60 and Jurkat cells induced with IFNγ. HL60 (A) and Jurkat cells (B) were treated or not [control (Ctrl)] with 50 µM EQ for 30 minutes at 37°C, then washed prior to incubation for 20 minutes with IFNγ. Washed cells were then analyzed at different time points as described in Materials and Methods. Results are the mean of three independent experiments; error bars indicate S.D. *P < 0.05, determined using Student’s t test as described under the Materials and Methods section. Western blot is representative of three independent experiments. Quantification of STAT1 phosphorylation in western blots was carried out using ImageJ software. a.u., arbitrary unit; N.I., no interferon; pSTAT1, phosphorylated STAT1.
Discussion
Etoposide is currently one of the most commonly used antitumor drugs. However, its use in clinics is associated with an increased risk of secondary leukemia (Pendleton et al., 2014). Several threads of experimental evidence indicate that EQ, a reactive metabolite of ETOP, is involved in ETOP-related leukemia through alteration of the topoisomerase II function (Gantchev and Hunting, 1998; Kagan et al., 1999; Fan et al., 2006; Jacob et al., 2011; Vlasova et al., 2011; Smith et al., 2014; Gibson et al., 2016). However, effects of EQ on other cellular targets could also contribute to ETOP-related leukemogenesis (Haim et al., 1987; Fan et al., 2006; Rojas et al., 2009). We investigated the possibility of protein tyrosine phosphatase TCPTP being a target of EQ. TCPTP plays a pivotal role in normal and malignant hematopoiesis through the negative regulation of the JAK/STAT signaling pathway (Dorritie et al., 2014). Interestingly, ETOP was reported to activate STAT1 signaling in HeLa cells. In addition, it has been shown that protein tyrosine phosphatases similar to TCPTP could be inhibited by polyaromatic quinones (Wang et al., 2004; Iwamoto et al., 2007). In line with this, we found in this work that EQ, the quinone metabolite of ETOP, was able to inhibit cellular TCPTP activity with subsequent increase in tyrosine phosphorylation of STAT1 in hematopoietic cell lines. Using further molecular and kinetic approaches, we show that EQ is an irreversible inhibitor of TCPTP. The second-order rate constant (kinact) for this inhibition was found to be ∼810 M−1⋅min−1 (Fig. 5B). This rate constant is close to values found for naphthoquinone-dependent inhibition of human protein tyrosine phosphatase 1B (420 M−1⋅min−1), a tyrosine phosphatase structurally related to TCPTP (Wang et al., 2004). Interestingly, naphthoquinones have been shown to alter cell signaling, in particular through increased tyrosine phosphorylation (Iwamoto et al., 2007; Klotz et al., 2014). In addition, the kinact value for the inhibition of TCPTP by EQ is comparable to that reported previously for hydrogen peroxide (600 M−1⋅min−1), a known endogenous regulator of tyrosine protein phosphatases (Seiner et al., 2007; Ostman et al., 2011). Inhibition of TCPTP by EQ was not reversed by buffer exchange (Fig. 4A). This observation, along with the time-dependent nature of the reaction (Fig. 5A), indicates that the inhibition of TCPTP by EQ is irreversible. It is well known that the activity of protein tyrosine phosphatases such as TCPTP relies on a reactive active site cysteine that can be modified by oxidants or electrophilic chemicals with subsequent loss of activity and altered cell signaling (Ostman et al., 2011; Klotz et al., 2014). In addition, several quinones, such as EQ, are Michael acceptors that can covalently react with cysteine residues to form irreversible adducts (Fan et al., 2006; Li et al., 2016; Bolton et al., 2018). Accordingly, we found that the irreversible nature of the inhibition of TCPTP by EQ is due to formation of covalent adducts on the catalytic cysteine residue of the enzyme (cysteine 216). This was confirmed by the fact that the inhibition process of TCPTP by EQ was slowed by addition of the competitive TCPTP inhibitor orthovanadate and by computational docking approaches. More importantly, mass spectrometry analysis of TCPTP inhibited by EQ shows that the active site cysteine residue of the enzyme is indeed adducted by EQ (Fig. 3C). Interestingly, recent experimental data indicate that EQ poisons topoisomerase II enzymes through cysteine adduction mechanisms, and that this covalent modification of topoisomerase II by EQ is likely to contribute to ETOP-related leukemogenesis (Jacob et al., 2011; Smith et al., 2014). Of note, the activity of EQ against topoisomerase IIα was found to be considerably higher than that of ETOP. Indeed, EQ can inhibit DNA relaxation at 100-fold lower levels (1 vs. 100 μM) of drug when compared with ETOP (Gibson et al., 2016). In their study on the inhibition of topoisomerase IIβ by EQ and ETOP, Smith et al. (2014) reported that whereas 15 μM EQ inhibited DNA cleavage by Topo2β in 6 minutes, less than 15% inhibition was observed with the parent drug ETOP. In addition, EQ is able to inhibit ATP hydrolysis by topoisomerase IIα, whereas ETOP cannot (Gibson et al., 2016). Interestingly, while EQ was found to inhibit TCPTP, the parent drug did not. This is in agreement with the electrophilic nature of EQ, which enables it to covalently react with the catalytic cysteine of TCPTP through Michael addition (Fan et al., 2006; Bolton et al., 2018). Tyrosine phosphatases closely related to TCPTP, such as PTP1B, have been shown to be inhibited by naphthoquinones through active site cysteine covalent modification (Iwamoto et al., 2007; Klotz et al., 2014). We found that EQ was also able to inhibit the phosphatase activity of PTP1B (Supplemental Fig. 4). Although we cannot rule out that EQ may affect STAT1 tyrosine phosphorylation through effects on other tyrosine phosphatases, small interfering RNA–mediated knockout of TCPTP in Jurkat cells and knockout mice models clearly indicates that impairment of TCPTP activity strongly impacts STAT1 tyrosine phosphorylation (Simoncic et al., 2002; Kleppe et al., 2010). Increasing evidence indicates that cytochrome P450/myeloperoxidase-dependent oxidative activation of ETOP into reactive metabolites (notably EQ) in hematopoietic cells is a key contributor to ETOP-related leukemia (Zhuo et al., 2004; Fan et al., 2006; Jacob et al., 2011; Vlasova et al., 2011; Smith et al., 2014; Gibson et al., 2016; Atwal et al., 2017). We show here that in addition to disruption of topoisomerase II functions, EQ could also contribute to ETOP-related leukemia through interactions with TCPTP and subsequent cell signaling alteration.
Acknowledgments
We thank the technical platform “BioProfiler-UFLC” (BFA Unit, Paris Diderot University) for provision of UFLC facilities. We are grateful to Dr. Oliver Brookes for English language proofreading of this manuscript.
Authorship Contributions
Participated in research design: Nian, Berthelet, Rodrigues Lima.
Conducted experiments: Nian, Berthelet, Bui, Ganesan, Leger.
Performed data analysis: Nian, Berthelet, Zhang, Bui, Liu, Duval, Ganesan, Leger, Chomienne, Busi, Guidez, Dupret, Rodrigues Lima.
Wrote or contributed to the writing of the manuscript: Nian, Berthelet, Xu, Ganesan, Leger, Busi, Rodrigues Lima.
Footnotes
- Received February 18, 2019.
- Accepted June 7, 2019.
This work was supported by University Paris Diderot and CNRS. Q.N., W.Z., and R.L. are supported by China Scholarship Council Ph.D. fellowships. J.B. was supported by a Ph.D. fellowship from Région Ile de France (Cancéropole 2015).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- DTT
- dithiothreitol
- EQ
- etoposide quinone
- ETOP
- etoposide
- FA
- formic acid
- FAM
- fluorescein amadite
- 5-IAF
- fluorescein 5-iodoacetamide
- IFNγ
- interferon γ
- JAK
- Janus kinase
- MPO
- myeloperoxidase
- MS
- mass spectrometry
- MS/MS
- tandem MS
- NBT
- nitroblue tetrazolium
- pNPP
- p-nitrophenyl phosphate
- PTP
- protein tyrosine phosphatase
- RP-UFLC
- reverse-phase ultrafast liquid chromatography
- STAT
- signal transducer and activator of transcription
- TCPTP
- T-cell protein tyrosine phosphatase
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}