


Visual Overview

Abstract
Developing small molecules that indirectly regulate Mcl-1 function has attracted a lot of attention in recent years. Here, we report the discovery of an aminopyrazole, 2-([1,1′-biphenyl]-4-yl)-N-(5-cyclobutyl-1H-pyrazol-3-yl)acetamide (analog 24), which selectively inhibited cyclin-dependent kinase (CDK) 5 over CDK2 in cancer cell lines. We also show that analog 24 reduced Mcl-1 levels in a concentration-dependent manner in cancer cell lines. Using a panel of doxycycline inducible cell lines, we show that CDK5 inhibitor 24 selectively modulates Mcl-1 function while the CDK4/6 inhibitor 6-acetyl-8-cyclopentyl-5-methyl-2-(5-(piperazin-1-yl)pyridin-2-ylamino)pyrido[2,3-day]pyrimidin-7(8H)-one does not. Previous studies using RNA interference and CRISPR showed that concurrent elimination of Bcl-xL and Mcl-1 resulted in induction of apoptosis. In pancreatic cancer cell lines, we show that either CDK5 knockdown or expression of a dominant negative CDK5 when combined with Bcl2 inhibitor results in synergistic induction of apoptosis. Moreover, concurrent pharmacological perturbation of Mcl-1 and Bcl-xL in pancreatic cancer cell lines using a CDK5 inhibitor analog 24 that reduced Mcl-1 levels and 4-(4-{[2-(4-chlorophenyl)-5,5-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-[(4-{[(2R)-4-(4-morpholinyl)-1-(phenylsulfanyl)-2-butanyl]amino}-3-[(trifluoromethyl)sulfonyl]phenyl)sulfonyl] benzamide (navitoclax), a Bcl-2/Bcl-xL/Bcl-w inhibitor, resulted in synergistic inhibition of cell growth and induction of apoptosis. In conclusion, we demonstrate targeting CDK5 will sensitize pancreatic cancers to Bcl-2 inhibitors.
SIGNIFICANCE STATEMENT Mcl-1 is stabilized by CDK5-mediated phosphorylation in pancreatic ductal adenocarcinoma, resulting in the deregulation of the apoptotic pathway. Thus, genetic or pharmacological targeting of CDK5 sensitizes pancreatic cancers to Bcl-2 inhibitors, such as navitoclax.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the deadliest form of pancreatic cancer and accounts for mortality among a majority of pancreatic cancer patients (Siegel et al., 2016). It is estimated that in 2019, 3.2% of all new cases and 7.5% of all cancer-related deaths will be due to pancreatic cancer (https://seer.cancer.gov/statfacts/html/pancreas.html). Limited therapeutic options as a result of late stage diagnosis have contributed to the poor clinical outcome, with only ∼7% of PDAC patients reaching the 5-year survival mark (Siegel et al., 2016). A common hallmark of cancer is evasion of cell death, which contributes to the persistence of pancreatic cancer and its resistance to chemotherapy (Westphal and Kalthoff, 2003). Elevated expression of antiapoptotic protein levels (Bcl-xL and Mcl-1) observed in PDAC suggests that the apoptotic pathway is deregulated (Campani et al., 2001; Evans et al., 2001) and targeting them is a viable therapeutic strategy for PDAC (Takahashi et al., 2013; Abulwerdi et al., 2014). Recent studies demonstrated that concurrent inactivation of Bcl-xL and Mcl-1 resulted in robust induction of apoptosis (Lopez et al., 2010; Rajule et al., 2012; O’Neill et al., 2016). As a result, Bcl-xL and Mcl-1 are considered attractive therapeutic targets for cancer therapy (Tse et al., 2008; Touzeau et al., 2011; Leverson et al., 2015; Contreras et al., 2018).
Abbott Laboratories (ABT) successfully developed direct inhibitors of Bcl-xL/Bcl-2/Bcl-w (the first drugs to target Bcl-2 family proteins): 4-{4-[(4′-chloro-2-biphenylyl)methyl]-1-piperazinyl}-N-[(4-{[(2R)-4-(dimethylamino)-1-(phenylsulfanyl)-2-butanyl]amino}-3-nitrophenyl) sulfonyl]benzamide (ABT-737); 4-(4-{[2-(4-chlorophenyl)-5,5-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-[(4-{[(2R)-4-(4-morpholinyl)-1-(phenylsulfanyl)-2-butanyl]amino}-3-[(trifluoromethyl)sulfonyl]phenyl)sulfonyl] benzamide [(ABT-263), navitoclax]; and 4-(4-{[2-(4-chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Tse et al., 2008; Touzeau et al., 2011). However, resistance to Bcl-xL/Bcl-2/Bcl-w inhibition has been attributed to compensatory activity by Mcl-1 (Konopleva et al., 2006; Mazumder et al., 2012). Consequently, Mcl-1 inactivation sensitizes cancer cells to Bcl-xL/Bcl-2/Bcl-w inhibitors (Peddaboina et al., 2012; Mohammad et al., 2017). While the development of direct Mcl-1 inhibitors is currently in preclinical and clinical development (AMG-176 [Spiro[5,7-etheno-1H,11H-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2′H)- naphthalen]-8(9H)-one, 6′-chloro-3′,4′,12,13,16,16a,17,18,18a,19-decahydro-16-methoxy-11,12-dimethyl- , 10,10-dioxide, (1′S,11R,12S,14E,16S,16aR,18aR)-], AZD5991 [(Z)-16-chloro-11,21,25,61-tetramethyl-11H,21H,61H-10-oxa-4,8-dithia-1(7,3)-indola-2(4,3),6(3,5)-dipyrazola-9(3,1)-naphthalenacyclotridecaphane-12-carboxylic acid], and S63845 [(R)-2-((5-(3-chloro-2-methyl-4-(2-(4-methylpiperazin-1-yl)ethoxy)phenyl)-6-(5-fluorofuran-2-yl)thieno[2,3-d]pyrimidin-4-yl)oxy)-3-(2-((1-(2,2,2-trifluoroethyl)-1H-pyrazol-5-yl)methoxy)phenyl)propanoic acid]) (Kotschy et al., 2016; Letai, 2016; Merino et al., 2017) targeting Mcl-1 indirectly is an alternate approach.
Several members of the cyclin-dependent kinases (CDKs) regulate the stability and expression of Mcl-1. Therefore, an alternate strategy to target Mcl-1 would be through modulation of members of the CDK family (MacCallum et al., 2005; Lowman et al., 2010; Choudhary et al., 2015b). Specifically, CDK5 regulates Mcl-1 function through phosphorylation of the endogenous Mcl-1 antagonist, NOXA (Lowman et al., 2010).
The kinase activity of CDK5 is regulated by p35, which has a myristoylation site at the N-terminus. In pancreatic tumors and cell lines, we previously showed the presence of elevated p25 levels, which is a cleaved product of p35 (Eggers et al., 2011). The cleavage of p35 to p25 leads to two changes: 1) mislocalization of CDK5/p25 to the cytoplasm due to loss of myristoylated anchoring by p35, and 2) increased half-life of CDK5/p25 (Patrick et al., 1999). These changes in CDK5 function/regulation along with the high homology observed between CDK2 and CDK5 indicate that in pancreatic cancer, CDK2 phosphorylation sites on Mcl-1 could now be targeted by CDK5/p25 (Kobayashi et al., 2007). Together, these studies suggest that a CDK5 inhibitor will disable Mcl-1 function and can be used in combination with Bcl-2/Bcl-xL/Bcl-w inhibitors as a promising therapeutic strategy for pancreatic cancer.
Small molecules containing the aminopyrazole core have shown promise as CDK inhibitors (Pevarello et al., 2004; Robb et al., 2017). Recent work from our laboratory characterized an aminopyrazole analog (CP-668863 [N-(3-cyclobutyl-1H-pyrazol-5-yl)-2-(naphthalen-2-yl)acetamide]) that inhibited tumor growth in a xenograft model of colorectal cancer (Robb et al., 2018). Using CP-668863 as a guide, we synthesized a focused library of aminopyrazole analogs and identified a potent CDK2/5 inhibitor, 2-([1,1′-biphenyl]-4-yl)-N-(5-cyclobutyl-1H-pyrazol-3-yl)acetamide (analog 24). We found that on an average (in five pancreatic cancer cell lines) analog 24 was ∼2-fold more potent than AT7519 [4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-1H-pyrazole-3-carboxamide] and ∼45-fold more potent than roscovitine [(2R)-2-[[6-(benzylamino)-9-propan-2-ylpurin-2-yl]amino]butan-1-ol] (Rana et al., 2018). Here, we show that analog 24 selectively inhibits the kinase activity of CDK5 over CDK2 in cancer cells and results in reduced Mcl-1 levels. We demonstrate selective perturbation of Mcl-1 function over Bcl-2/Bcl-xL/Bcl-w by analog 24 in cells using a chemical genetic study. Moreover, in pancreatic cancer cell lines, CDK5 knockdown or analog 24 treatment synergistically induced apoptosis when combined with the Bcl-2 inhibitor navitoclax.
Materials and Methods
Reagents and Antibodies: Inhibitors.
ABT-263; ABT-737; 4-(4-{[2-(4-chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide; 4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-1H-pyrazole-3-carboxamide; 5-[3-[4-(aminomethyl)phenoxy]propyl]-2-[(8E)-8-(1,3-benzothiazol-2-ylhydrazinylidene)-6,7-dihydro-5H-naphthalen-2-yl]-1,3-thiazole-4-carboxylic acid (WEHI-539); and 6-acetyl-8-cyclopentyl-5-methyl-2-(5-(piperazin-1-yl)pyridin-2-ylamino)pyrido[2,3-day]pyrimidin-7(8H)-one (palbociclib) were purchased from Selleckchem. Results on analog 24 were reported previously by our laboratory (Rana et al., 2018). Antibodies P-RB S807/811 (9308), RB (9309), tubulin (3873), Bcl-xL (2762), Mcl-1 (5453), poly ADP ribose polymerase [(PARP) 9542], Cl. PARP (9541), CDK5 (2506), P-AKT S473 (4060), Akt (9272), and CDK12 (11973) were purchased from CST. P-FAK S732 (ab4392) and FAK (ab4792) were purchased from Abcam. Horseradish peroxidase–conjugated goat anti-mouse IgG (G-21040) and anti-rabbit IgG (32460) secondary antibodies were purchased from Thermo Fisher Scientific.
Pancreatic Cell Lines.
S2-013 cells were cultured in RPMI 1640 medium (#SH30027.01; HyClone). MIA PaCa-2 cells were cultured in Dulbecco’s modified Eagle’s medium high-glucose medium (#SH30022.01; HyClone). S2013 cells expressing CDK5 dominant-negative (CDK5DN) (generated using vectors kindly provided by Barry Nelkin, Johns Hopkins University School of Medicine, Baltimore, MD) (Eggers et al., 2011) were cultured in RPMI 1640 medium. All cell lines were supplemented with 10% FBS (#26140-079; Gibco by LifeTechnologies) and 1% penicillin-streptomycin (#SV30010; HyClone) and cultured at 5% CO2 at 37°C.
HeLa Cell Lines.
Doxycycline (Dox) inducible HeLa cells (HeLa-Dox-GFP, HeLa-Dox-Bad3SA, and HeLa-Dox-Noxa cells) (Lopez et al., 2010) were cultured in Dulbecco’s modified Eagle’s medium high-glucose medium (#SH30022.01; HyClone) supplemented with 10% FBS (#26140-079; Gibco by LifeTechnologies) and 1% penicillin-streptomycin (#SV30010; HyClone) and cultured at 5% CO2 at 37°C. Cells were treated with 1 µg/ml of doxycycline to induce the expression of GFP, BAD3SA, or Noxa.
Western Blot Analyses.
Cells were washed with cold 1X PBS and scraped before being lysed by a buffer comprised of 50 mM Tris, 100 mM sodium chloride, 1% NP40, 2 nM EDTA, 20% SDS, 1 mM sodium orthovandate, 25 mM sodium fluoride, β-glycerophosphate, and 1 mmol/l phenylmethylsulfonyl fluoride. Samples were incubated on ice for 30 minutes and vortexed in 15-minute increments. Samples were centrifuged at 14,000g for 10 minutes at 4°C and supernatant was collected. Protein quantification was determined by BCA Protein Assay (#23225; Pierce). Then, 20–40μg protein samples were run on 4%–15% gradient gels (BioRad) in 1X Tris-Glycine-SDS Buffer (#T32080; Research Products International Corporation) at 90 V for ∼90 minutes and separated by SDS-PAGE. Samples were transferred to a polyvinylidene difluoride membrane by the semidry transfer method (ThermoScientific #35035) and run at 18 V for ∼35 minutes. Membranes were blocked in 5% milk diluted in 1X Tris-buffered saline with 0.1% Tween (1X Tris-buffered saline/Tween 20) for 1 hour at room temperature, rocking at low speed. Primary antibodies were diluted in 5% milk in 1X Tris-buffered saline/Tween 20 and rocked gently overnight at 4°C. Membranes were incubated with the appropriate horseradish peroxidase–conjugated secondary antibody for 1 hour at room temperature while gently rocking. Three washes (10 minutes) with 1X Tris-buffered saline/Tween 20 occurred before and after secondary antibody. ECL Prime (#RPB2236; GE Healthcare) was used to detect protein expression.
Caspase Assay.
Cells were plated in a 384-well white-walled, clear-bottom plate at 2500 cells per well in 100 µl and subjected to 4.5 µM doxycycline. Cells were incubated overnight to adhere to the plate, and were then treated at indicated concentrations. After cells were treated and the plate was incubated for the indicated time, Presto-blue was added at a volume of 1/10 total volume per well and cells were incubated for 10 minutes. The plate was then read for fluorescence at 560excitation/590emission. Caspase-Glo (Promega G8093) reagent was added and the plate was incubated for 30 minutes. The plate was then read for luminescence at 1000 milliseconds. Values were calculated as ([luminescence × 100]/fluorescence)/DMSOaverage.
Cell Viability Assay.
Cells were plated at 4000 cells/well in a 96-well plate and allowed to adhere overnight. On the following day, cells were treated with compounds individually or in combination (2-fold dilutions starting at 100 μM). PrestoBlue reagent (#A13262; Invitrogen) was added to cells after 72-hour drug incubation to assess the growth inhibition (Radhakrishnan et al., 2013). Fluorescence excitation/emission was measured at 560/590 nM using the SpectraMax M5e instrument. Growth inhibition was calculated using 100 − [100 × (samples − T0)/(T100 − T0)], where T0 is the vehicle control reading immediately following drug addition, and T100 is the control reading at the end of the 72-hour incubation.
Small Interfering RNA Transfection.
S2013 and MIA PaCa-2 cells were seeded at a density of 2.5 × 105 cells/well in a six-well plate and incubated overnight to allow them to adhere to the plate. Opti-MEM (Gibco) was used for all transfection experiments. Cells were transiently transfected with 160 nM of appropriate antisense oligonucleotides [Sc-29263 for the small interfering CDK5 pool of three target-specific small interfering RNAs (siRNAs) and Sc-37007 for scrambled control; SantaCruz] using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol. Cells were then incubated for 48 hours and 10 µM of ABT-263 was added to the wells wherever indicated for 6 hours. Cells were then harvested and subjected to western blot analysis.
Dominant Negative CDK5 Cell Line Study.
S2013 CDK5DN cells (Eggers et al., 2011) were seeded at a density of 1 × 106 cells in a 10-cm plate. Cells were treated with and without 3 µg/ml of doxycycline and incubated for 48 hours, followed by addition of 10 µM of ABT-263 to wells wherever indicated for 6 hours. Cells were then harvested and subjected to western blot analysis.
The Cancer Genome Atlas and Genotype-Tissue Expression Analyses.
The Cancer Genome Atlas (TCGA) provisional data were downloaded from the University of California Santa Cruz Xena web site (https://xenabrowser.net/datapages/) on January 8, 2019. The mRNA expressions (RNA sequences) of MCL-1 and Bcl-2 were analyzed in the pancreatic adenicarcinoma data set (pancreatic cancer − TCGA provisional; mean normalized). The genotype-tissue expression (GTEx) pancreatic samples were downloaded from the University of California Santa Cruz Xena web site (https://xenabrowser.net/heatmap/) on May 13, 2019. We assumed that the TCGA and GTEX samples came from a population with Gaussian distribution; therefore, we reported S.D. and used a t test.
Sample Size and Statistical Analyses.
Unless otherwise stated, all experiments were done at least as biologic duplicates with technical triplicates. The parameters reported are average ± S.D. Graphs and figures were generated using SigmaPlot 11.0 and Graphpad Prism statistical software (Graphpad Software, Inc.). Student’s t test (two-tailed) was used to determine significance between two groups, where P < 0.05 was considered significant (all reported P values are not hypothesis testing but descriptive only). Combination index (CI) values (Bryant et al., 2012) were determined by CalcuSyn 2.11.
Results
Cell-Based Studies Identified Analog 24 as a Selective CDK5 Inhibitor.
We, and others, have previously reported aminopyrazoles as CDK inhibitors with antitumor activities (Pevarello et al., 2004; Rana et al., 2018). A systematic structure-activity relationship study identified analog 24 as a potent CDK inhibitor (Rana et al., 2018). Cell-free kinase assays show that analog 24 is a CDK2/5 inhibitor (Fig. 1A). To test whether this holds true in a cellular assay, we evaluated analog 24 for its ability to inhibit CDK2 and CDK5 in MIA PaCa-2 and HeLa cells (Fig. 1B). We used previously reported CDK2 and CDK5 substrates, i.e., pRB (Ser807/811) and pFAK (Ser732), respectively (Knudsen and Wang, 1996; Xie et al., 2003; Romano and Giordano, 2008; Byth et al., 2009; Siemeister et al., 2012), as readouts to assess the ability of analog 24 to inhibit the corresponding CDKs. MIA PaCa-2 and HeLa cells treated with analog 24 showed a concentration-dependent decrease in the levels of pFAK (Ser732), suggesting inhibition of the kinase activity of CDK5. We observed some reduction in the levels of pRB at the 10 μM concentration; however, we did not see a clear concentration-response relationship, suggesting that analog 24 does not inhibit CDK2 in MIA PaCa-2 or HeLa cells.

(A) Chemical structure of aminopyrazole analog 24 and its cell-free kinase activity. (B) Concentration-dependent studies with analog 24 in MIA PaCa-2 and HeLa cells treated for 6 hours to assess its activity against CDK2 and CDK5 using western blot analyses. (C) Concentration-response study to determine the cellular CDK5 IC50 value for analog 24 in MIA PaCa-2 cells. (D) Concentration-dependent studies with analog 24 in MIA PaCa-2 and HeLa cells treated for 6 hours to assess its activity against Bcl-xL and Mcl-1 using western blot analyses. Presented blots are representative of at least two independent experiments.
Next, to estimate the cellular CDK5 IC50 value for analog 24, we subjected MIA PaCa-2 cells to increasing concentrations of analog 24 (0–5 μM) and incubated the treated cells for 12 hours. Quantification of pFAK levels from the immunoblot followed by curve fitting yielded a cellular CDK5 IC50 value of ∼1.1 μM for analog 24 (Fig. 1C). Although cell-free kinase assay suggested analog 24 as a CDK2/5 inhibitor, cell-based studies established analog 24 as a selective CDK5 inhibitor.
A previous report indicated that CDK5-mediated NOXA phosphorylation leads to the formation of a Mcl-1 survival complex and inhibition of CDK5 will likely drive the degradation of Mcl-1 (Lowman et al., 2010). To determine if analog 24 affected Mcl-1 levels, we probed the membranes with MIA PaCa-2 and HeLa lysates with Mcl-1 and Bcl-xL antibodies. We observed a concentration-dependent decrease in Mcl-1 but not Bcl-xL levels. This suggests that CDK5 inhibition by analog 24 also leads to downregulation of Mcl-1 levels (Fig. 1D).
Chemical Genetic Studies Show that Analog 24 Selectively Perturbs Mcl-1 Function.
Using a panel of doxycycline-inducible HeLa cell lines (Fig. 2), (Lopez et al., 2010; Chen et al., 2011; Rajule et al., 2012) we explored the effects of analog 24 on the antiapoptotic pathway proteins. Briefly, the HeLa-Dox-GFP cell line in the presence of Dox expresses GFP, and therefore has functional Bcl-2/Bcl-xL/Bcl-w and Mcl-1 (Fig. 2, left). The HeLa-Dox-Noxa cell line in the presence of Dox expresses Noxa, which binds to and inactivates Mcl-1 but has functional Bcl-2/Bcl-xL/Bcl-w (Fig. 2, middle). The HeLa-Dox-Bad3SA cell line in the presence of Dox expresses Bad3SA, which binds to and inactivates Bcl-2/Bcl-xL/Bcl-w but has functional Mcl-1 (Fig. 2, right).

Chemical genetic screening strategy to determine selective inactivation of Mcl-1 by analog 24 using doxycycline-inducible cell lines.
We first screened a well-characterized Bcl-2 inhibitor (ABT-263, also known as navitoclax) in the three Dox-inducible cell lines (Fig. 3A). We observed a concentration-dependent increase in caspase 3/7 activity only in the Dox-Noxa cell lines because Dox treatment induced expression of Noxa, which inactivates Mcl-1 and ABT-263 inhibits Bcl-2/Bcl-xL/Bcl-w (Fig. 3A). Next, we subjected the three cell lines to analog 24 and conducted a time-course (Fig. 3B) and concentration-response (Fig. 3C) study. Remarkably, we observed increased caspase 3/7 activity only in the HeLa-Dox-Bad3SA cell line and not the HeLa-Dox-GFP or HeLa-Dox-Noxa cells in both the concentration- and time-dependent treatments. This suggests that analog 24 selectively perturbs Mcl-1 function.

Chemical genetic screens with caspase 3/7 assay readout. Studies in HeLa-Dox cell lines: (A) concentration-response results with ABT-263 (n = 3, ±S.D.); (B) time course with analog 24 (n = 3, ±S.D.). (C) Concentration-response results with analog 24 (n = 3, ±S.D.). (D) Western blot analyses of concentration-response studies in HeLa-Dox cell lines with analog 24 and palbociclib. Blots are representative of at least two independent experiments. (E) Concentration-response studies in HeLa-GFP cells treated for 6 hours with analog 24 and ABT-263 individually and in combination (n = 3, ±S.D.). (F) Concentration-response studies in HeLa-GFP cells treated for 6 hours with palbociclib and ABT-263 individually and as a combination (n = 3, ±S.D.).
To confirm that the selective induction of caspase 3/7 in the HeLa-Dox-Noxa cell line by analog 24 is a result of Mcl-1 downregulation, we performed western blot analyses of the lysates from a concentration-response study with analog 24 in all three HeLa-Dox cell lines (Fig. 3D). We observed a concentration-dependent decrease in Mcl-1 levels in each of the three HeLa-Dox cell lines (Fig. 3D, top panel). However, PARP cleavage, a hallmark of apoptosis, was only observed in the HeLa-Bad3SA cell line. To determine if this effect was CDK5 selective we conducted the same study with a CDK4/6 selective inhibitor, palbociclib. We observed no changes in levels of Mcl-1 or PARP cleavage in all three HeLa-Dox cell lines treated with palbociclib (Fig. 3D, bottom panel). Together, these results show that analog 24 inhibits CDK5 and as a consequence perturbs Mcl-1 function.
Analog 24 Synergistically Induced Apoptosis When Combined with ABT-263.
Genetic knockdown and knockout studies demonstrated that concurrent elimination of Bcl-xL and Mcl-1 induced apoptosis (Lopez et al., 2010; O’Neill et al., 2016). To determine if this extends to pharmacological perturbations we subjected HeLa-GFP cells to increasing concentrations of analog 24 or ABT-263 or the combination and assessed the effects using caspase 3/7 assay (Fig. 3E). Under the assay conditions, we observed induction of apoptosis only in the combination treatment. Importantly, no such effect was observed with the CDK4/6 inhibitor, palbociclib, and ABT-263 combination (Fig. 3F). Together, these studies show that concurrent pharmacological inactivation of Bcl-xL and Mcl-1 synergistically induced apoptosis.
Combining 24 with the ABT Compounds Synergistically Induced Apoptosis and Inhibited Growth in Pancreatic Cancer Cell Lines.
Next, we determined if the observed synergism would extend to pancreatic cancer cell lines. In a concentration-response study, the pancreatic cancer cell lines MIA PaCa-2 and S2-013 were treated individually with ABT-737, ABT-263, or analog 24, or the combination of ABT compounds + analog 24 (Fig. 4). The induction of apoptosis was monitored by measuring caspase 3/7 levels following 6-hour incubation. In both cell lines we observed a robust concentration-dependent increase in caspase 3/7 levels with the combination of ABT + analog 24. This suggests synergism between analog 24 and the ABT compounds in pancreatic cancer cell lines.

Combination studies in pancreatic cancer cell lines with caspase 3/7 assay readout. (A) Concentration-response studies with analog 24 and ABT-263 individually and as a combination in the MIA PaCa-2 cell lines (n = 3, ±S.D.). (B) Analog 24 and ABT-737 individually and as a combination in the MIA PaCa-2 cell lines (n = 3, ±S.D.). (C) Analog 24 and ABT-263 individually and as a combination in the S2-013 cell lines (n = 3, ±S.D.). (D) Analog 24 and ABT-737 individually and as a combination in the S2-013 cell lines (n = 3, ±S.D.).
We then treated S2013 cells individually with ABT-737, ABT-263, or analog 24, or the combination of ABT compounds + analog 24 and the lysates were probed for the levels of Mcl-1, PARP, and cleaved PARP by western blot analyses. Consistent with the data shown in Fig. 1, we observed a reduction in Mcl-1 levels in the analog 24–treated cells and induction of PARP cleavage was only observed with the combined treatment of analog 24 and ABT compounds (Fig. 5A). We conducted similar experiments with two additional control compounds, a CDK 5/9 inhibitor [4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-1H-pyrazole-3-carboxamide] and a Bcl-xL selective inhibitor (WEHI-539) and observed similar effects. We concluded from these studies that concurrent inactivation of Mcl-1 (indirectly) and Bcl-xL/Bcl-2/Bcl-w results in synergistic induction of apoptosis (Fig. 5B).

Synergism studies in pancreatic cancer cell lines. (A) Western blot analyses of cleaved PARP and Mcl-1 levels in S2-013 cells treated with ABT compounds, analog 24, or the combination for 12 hours. (B) Combination studies of 4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-1H-pyrazole-3-carboxamide (AT7519), analog 24, and palbociclib with ABT-263 and WEHI-539 in MIA PaCa-2 cells treated for 6 hours. (C) CDK5 knockdown (pooled siRNA) and synergism studies with ABT-263 in S2-013 and MIA PaCa-2 cells. (D) Effect of ABT-263 in S2013 CDK5DN doxycycline-inducible cells. For all of the aforementioned experiments, the presented blots are representative of at least two independent experiments. (E) Dose-effect curve for analog 24 and ABT-263 individually and in combination in MIA PaCa-2 cells. (F) Tables summarizing the median-effective dose (Dm) for analog 24 and ABT-263 individually and in combination in MIA PaCa-2 cells, along with effective dose (ED) and CI values derived from growth inhibition studies in MIA PaCa-2 cells (n = 2); a CI value of 0.7–1.0 is considered mildly synergistic, a CI value of 0.7–0.3 is considered moderately synergistic, and a CI value <0.3 is considered strongly synergistic.
Since analog 24 inhibits CDK5, which we believe is the reason for the inactivation of Mcl-1 leading to the observed synergism with navitoclax, we genetically validated this by CDK5 knockdown using siRNA in the pancreatic cancer cell lines MIA PaCa-2 and S2013. Figure 5C shows that 48 hours post CDK5 siRNA (pool of three target-specific siRNAs) transfection results in robust knock down of CDK5 while no change was observed in CDK12 levels. Treatment of CDK5 siRNA-transfected MIA PaCa-2 and S2013 cells with ABT-263 (navitoclax) for 6 hours showed elevated levels of cleaved PARP in the CDK5 siRNA-transfected cells when compared with control or scrambled siRNA cells (Fig. 5C). These results show that knock down of CDK5 sensitizes pancreatic cancer cells to navitoclax-induced PARP cleavage. We also validated these results by using doxycycline-inducible CDK5DN S2013 cells (Fig. 5D). Treatment of S2013 CDK5DN cells with ABT-263 showed elevated levels of cleaved PARP in the doxycycline-induced cells compared with ABT-263–treated noninduced cells, suggesting that inhibition of CDK5 function indeed plays an important role in sensitizing pancreatic cancer cells to navitoclax-induced PARP cleavage. Expression of CDK5DN resulted in reduced FAK phosphorylation but no change was observed in Akt phosphorylation.
To determine whether the pharmacological inhibition of CDK5 and Bcl-xL/Bcl-2/Bcl-w will synergistically inhibit growth of pancreatic cancer cell lines, MIA PaCa-2 cells were treated with increasing concentrations of analog 24 or navitoclax, or the combination. The IC50 values were derived from growth curves (Fig. 5E) and used to calculate the median concentration effect of analog 24, navitoclax, or the combination and the CI values (Fig. 5F) using CalcuSyn (Chou, 2006; Bryant et al., 2012). The study revealed that the median effective concentration for the combination (analog 24 + navitoclax) is lower than the individual treatments (Fig. 5F, top table). Consistently, treatment of analog 24 and ABT-263 was indeed synergistic in MIA PaCa-2 (averageCI = 0.36) cells at high effective concentrations (effective doses of 75, 90, and 95) (Fig. 5F, bottom table). (Chou, 2006) This observation is consistent with reported studies that show apoptosis only occurs when both the Mcl-1 and Bcl-xL/Bcl-2/Bcl-w arms are concurrently disabled (Lopez et al., 2010; Rajule et al., 2012; Contreras et al., 2018).
Model for a Previously Unexplored Therapeutic Strategy for Pancreatic Cancer.
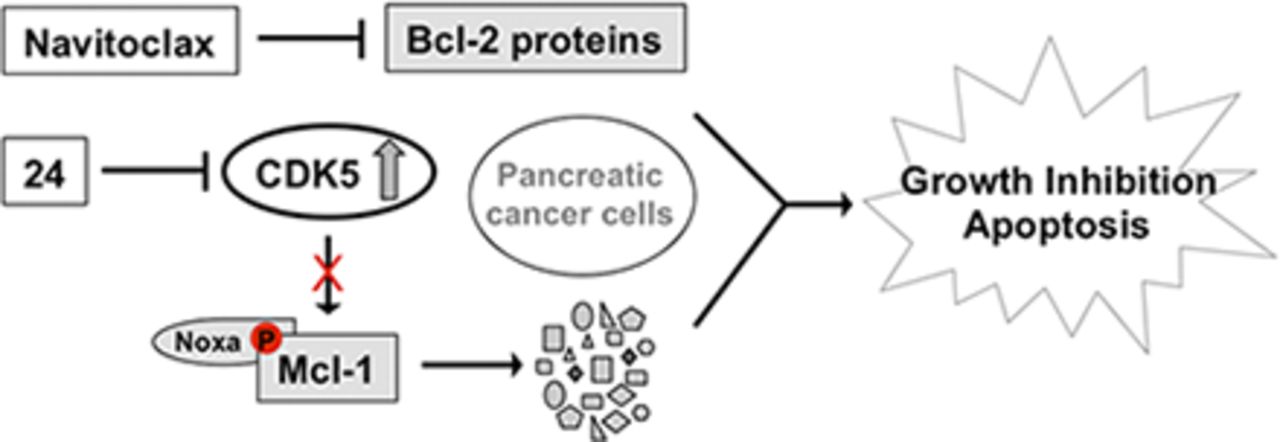
Analyses of the TCGA and GTEX data revealed that Mcl-1 mRNA levels are nearly identical in normal pancreas tissue and pancreatic cancer (Fig. 6A). It is important to note that among the Bcl-2 family of proteins only Bcl-xL mRNA levels are elevated in the primary tumors when compared with normal tissue (Fig. 6, B–D). Reported studies in pancreatic tumors have shown elevated Mcl-1 protein levels (Chen et al., 2013), suggesting post-translational stabilization of Mcl-1. There are several reported studies that provide mechanistic basis for the post-translational stabilization of Mcl-1 such as phosphorylation of key residues in the proline glutamic acid serine threonine domain of Mcl-1 (Domina et al., 2004). Consistent with elevated levels of Mcl-1 in pancreatic tumors, we observed a similar trend in a panel of pancreatic cancer cell lines when compared with human pancreatic nestin expressing cells (Fig. 6E). Together, these results suggest that Mcl-1 levels are elevated in pancreatic cancer cell lines probably due to post-translational modifications, such as phosphorylation, which stabilize Mcl-1. Our model argues that CDK5-mediated stabilization of Mcl-1 drives oncogenesis, consistent with elevated levels of CDK5 observed in pancreatic cancer cell lines and tumors (Eggers et al., 2011). Here, we propose that concurrent inactivation of Bcl-2 proteins by direct inhibitors and Mcl-1 through CDK5 inhibition is an attractive therapeutic strategy for pancreatic cancer (Fig. 6F).

Rationale for using analog 24 in pancreatic cancer therapy. (A–D) Analyses of TCGA and GTEx data, mRNA Mcl-1, Bcl-w, Bcl-xL, and Bcl-2 levels in normal pancreas tissue and pancreatic cancer. (E) Basal levels of Mcl-1 in human pancreatic nestin expressing (HPNE) and a panel of pancreatic cancer cell lines. Blots are representative of at least two independent experiments. (F) Our model for combining analog 24 and navitoclax, a Food and Drug Administration–approved Bcl-xL/Bcl-2/Bcl-w inhibitor, in pancreatic cancer therapy.
Discussion
Targeting antiapoptotic proteins to sensitize cancer cells to apoptosis has been extensively explored for the past two decades. Abbott Laboratories pioneered the translation of Bcl-2 inhibitors to the clinic by successfully developing direct inhibitors (ABT-737, ABT-263, and 4-(4-{[2-(4-chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide) that target Bcl-2 family proteins (Tse et al., 2008; Touzeau et al., 2011). Several studies have shown that Mcl-1 compensates for the loss of Bcl-xL/Bcl-2/Bcl-w to circumvent apoptosis, suggesting concurrent inactivation of Mcl-1 and Bcl-xL/Bcl-2/Bcl-w is necessary to induce apoptosis (Konopleva et al., 2006; van Delft et al., 2006; Chen et al., 2007; Choudhary et al., 2015a). For example, in colorectal cancer, treatment with the Bcl-xL inhibitor ABT-737 resulted in increased Mcl-1 expression (Peddaboina et al., 2012). Although direct Mcl-1 inhibitors are currently in preclinical and clinical development, (Kotschy et al., 2016; Letai, 2016; Merino et al., 2017) none have been approved by the Food and Drug Administration, suggesting the need for alternate approaches to perturb the Mcl-1 pathway.
CDK inhibitors have been used to indirectly modulate Mcl-1 function. The activity and stability of CDK5, an atypical CDK, is regulated by its binding partner p35. We previously reported elevated levels of p25, a cleaved form of p35, in pancreatic tumors and cell lines (Eggers et al., 2011). The CDK5/p25 complex is mislocalized to the cytoplasm and is more stable than CDK5/p35 (Patrick et al., 1999). CDK5 has been validated as a target in pancreatic cancer (Feldmann et al., 2010); however, the exact mechanism of how CDK5 could promote tumorigenesis is yet to be fully elucidated. Recently, we identified an aminopyrazole-based analog 24 that inhibited CDK2 and CDK5 with nanomolar potency (Rana et al., 2018). Here, we show that analog 24 is a selective CDK5 inhibitor with an IC50 value of ∼1.1 μM in MIA PaCa-2 cells (Fig. 1). Lowman et al. (2010) showed that CDK5 phosphorylates Noxa, a Mcl-1 antagonist, and thus allowed Mcl-1 to retain its antiapoptotic activity. Therefore, here we evaluated the effect of analog 24 on Mcl-1 levels in MIA PaCa-2 and HeLa cells (Fig. 1). Analog 24 reduced Mcl-1 levels in a concentration-dependent manner in both cell lines.
Using a panel of doxycycline-inducible cell lines, we characterized the selectivity of analog 24 for the Mcl-1 arm over the Bcl-xL/Bcl-w/Bcl-2 arm of the apoptotic pathway (Fig. 2). We screened analog 24 in a panel of three dox-inducible cell lines that either had functional Mcl-1 (HeLa-Dox-Bad3SA) or functional Bcl-xL/Bcl-w/Bcl-2 (HeLa-Dox-Noxa) or functional Mcl-1 and Bcl-xL/Bcl-w/Bcl-2 (HeLa-Dox-GFP). We anticipated one of three possible outcomes: 1) analog 24 is not selective, in which case we will observe caspase 3/7 activation in the HeLa-Dox-GFP cell line; 2) analog 24 is a Bcl-xL/Bcl-2/Bcl-w pathway inhibitor, in which case we will observe caspase 3/7 activation in the HeLa-Dox-Noxa cell line; or 3) analog 24 is a Mcl-1 pathway inhibitor, in which case we will observe caspase 3/7 activation in the HeLa-Dox-Bad3SA cell line.
The robustness of the assay was validated using the Bcl-xL/Bcl-2/Bcl-w inhibitor navitoclax (ABT-263). ABT-263 induced caspase activation in HeLa-Dox-Noxa cell lines, wherein dox expressed Noxa that binds and inactivates the Mcl-1 arm and ABT compounds inhibit the Bcl-xL/Bcl-2/Bcl-w arm, which resulted in caspase 3/7 activation (Fig. 3A). Under similar conditions, analog 24 induced caspase activation only in the HeLa-Dox-Bad3SA cell line, indicating selective inhibition of the Mcl-1 arm of the apoptotic pathway (Fig. 3, B and C). This is because in HeLa-Dox-GFP cells analog 24 inactivates Mcl-1; however, functional Bcl-xL/Bcl-2/Bcl-w protects cells from the induction of apoptosis. In the HeLa-Dox-Noxa cells analog 24 inactivates Mcl-1, leaving functional Bcl-xL/Bcl-2/Bcl-w to protect cells from the induction of apoptosis. On the other hand, in HeLa-Dox-Bad3SA cells, dox induction results in Bad expression, which inactivates Bcl-xL/Bcl-2/Bcl-w, while analog 24 inactivates Mcl-1, resulting in the induction of apoptosis.
We used PARP cleavage as a secondary readout to demonstrate the ability of analog 24 to reduce Mcl-1 levels and induce apoptosis. In all three dox-inducible cell lines, in the presence of analog 24, we observed reduction in Mcl-1 levels. However, analog 24 induced PARP cleavage only in the HeLa-Bad3SA cell line, demonstrating that concurrent inactivation of Mcl-1 and Bcl-xL/Bcl-2/Bcl-w is critical for PARP cleavage (Fig. 3D). No such PARP cleavage was observed with palbociclib (a selective CDK4/6 inhibitor) under similar conditions (Fig. 3D). Next, we hypothesized that a combination of analog 24 and ABT-263 will induce robust caspase activation in HeLa-Dox-GFP cell lines. Indeed, analog 24 synergized with ABT-263 and induced caspase 3/7 activity (Fig. 3E). Since analog 24 is a CDK5-selective inhibitor that inactivates Mcl-1, we used palbociclib, a CDK4/6 inhibitor, in combination with ABT-263 (Fig. 3F) as a negative control and observed no activation of caspase 3/7.
To determine if the observed synergism between the CDK5 inhibitor analog 24 and ABT-263 extends to pancreatic cancer cell lines, we treated MIA PaCa-2 and S2013 cells with analog 24, ABT-263, and ABT-737 individually or in combination in a concentration-response study and monitored the caspase 3/7 activity. Indeed, we observed synergistic induction of caspase 3/7 with the CDK5 inhibitor and ABT combination (Fig. 4). This suggests that CDK5 inhibition sensitizes pancreatic cancer cell lines to Bcl-2 inhibitors ABT-737 and navitoclax.
Next, we explored synergism in pancreatic cancer cell lines by monitoring PARP cleavage, which is another marker for the induction of apoptosis. Consistent with the caspase 3/7 results, only the combined treatment of analog 24, and ABT-263 or WEHI-539 (a Bcl-xL selective inhibitor) showed PARP cleavage. No PARP cleavage was observed in single treatments or when ABT-263 and WEHI-539 were combined with palbociclib (Fig. 5, A and B). To genetically validate sensitization of pancreatic cancer cell lines by the CDK5 inhibitor, we knocked down CDK5 using siRNA and doxycycline-inducible CDK5DN pancreatic cancer cell lines. In pancreatic cancer cell lines, where CDK5 level or CDK5 function was genetically manipulated, navitoclax robustly induced PARP cleavage in cells with CDK5 dysfunction when compared with the corresponding controls (Fig. 5, C and D). These studies demonstrate that targeting CDK5 perturbs the Mcl-1 arm; as a consequence, pancreatic cancer cell lines are sensitive to navitoclax-induced apoptosis.
A follow-up growth inhibition study in MIA PaCa-2 cells using analog 24 and navitoclax demonstrated that treatment of analog 24 and ABT-263 was synergistic as evidenced by averageCI values <0.4. A CI value of <1.0 for a given combination treatment suggests synergism. A CI value between 0.3 and 0.7 indicates moderate synergism. The growth inhibition studies suggest that analog 24 and navitoclax exhibit moderate synergism in a pancreatic cancer cell growth inhibition assay.
Analyses of the TCGA and GTEx data show that the mRNA levels of Bcl-2, Bcl-w, and Mcl-1 are lower in primary tumors compared with normal tissue, while Bcl-xL is elevated in primary tumors compared with normal tissue (Fig. 6, A–D). This indicates that Bcl-xL inhibitors are a viable option for pancreatic cancer therapy. However, previous studies have shown that inhibition of Bcl-xL results in elevated Mcl-1 levels (Konopleva et al., 2006; Mazumder et al., 2012). Moreover, the basal Mcl-1 protein levels are elevated in pancreatic cancer cell lines (Fig. 6E). (Miyamoto et al., 1999) This incongruent observation in the mRNA and protein levels of Mcl-1 suggests post-translational stabilization of Mcl-1 (Liu et al., 2005).
Since CDK5 (Eggers et al., 2011) and Mcl-1 (Fig. 6E) levels are elevated in pancreatic cancer cell lines and tumors (Chen et al., 2007), inhibitors of CDK5 such as analog 24 downregulate Mcl-1 and synergistically induce apoptosis when combined with Bcl-2 inhibitors. This therapeutic combination can be explored for the treatment of pancreatic cancer (Fig. 6F).
In conclusion, the study presented here characterized the aminopyrazole analog 24 as a CDK5-selective inhibitor. We also show that either genetic or pharmacological inhibition (analog 24) of CDK5 selectively perturbs the Mcl-1 arm of the apoptotic pathway. In pancreatic cancer cell lines, we observed synergistic inhibition of growth and induction of apoptosis when analog 24 was combined with navitoclax. Preclinical studies to extend the aforementioned findings to in vivo models are currently underway, and the results from these studies will be reported in due course.
Authorship Contributions
Participated in research design: Luo, Hollingsworth, Natarajan.
Conducted experiments: Kour, Rana, Contreras, King, Robb, Sonawane, Crawford, Barger, Kizhake, Bendjennat.
Performed data analysis: Kour, Rana, Contreras, King, Natarajan.
Wrote or contributed to the writing of the manuscript: Kour, Rana, Contreras, Natarajan.
Footnotes
- Received April 5, 2019.
- Accepted July 23, 2019.
↵1 S.Ko., S.R., and J.I.C. contributed equally to this work.
This work was supported in part by the National Institutes of Health [Grants CA182820, CA197999, CA054807, CA009476, CA205496, CA036727, and GM118437] and fellowships from the University of Nebraska Medical Center (to J.I.C. and S.Ko.).
Abbreviations
- ABT
- Abbott Laboratories
- ABT-263
- (navitoclax) 4-(4-{[2-(4-chlorophenyl)-5,5-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-[(4-{[(2R)-4-(4-morpholinyl)-1-(phenylsulfanyl)-2-butanyl]amino}-3-[(trifluoromethyl)sulfonyl]phenyl)sulfonyl] benzamide
- ABT-737
- 4-{4-[(4′-chloro-2-biphenylyl)methyl]-1-piperazinyl}-N-[(4-{[(2R)-4-(dimethylamino)-1-(phenylsulfanyl)-2-butanyl]amino}-3-nitrophenyl) sulfonyl]benzamide
- AMG-176
- spiro[5,7-etheno-1H,11H-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2′H)- naphthalen]-8(9H)-one, 6′-chloro-3′,4′,12,13,16,16a,17,18,18a,19-decahydro-16-methoxy-11,12-dimethyl- , 10,10-dioxide, (1′S,11R,12S,14E,16S,16aR,18aR)-
- AT7519
- 4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-1H-pyrazole-3-carboxamide
- analog 24
- 2-([1,1′-biphenyl]-4-yl)-N-(5-cyclobutyl-1H-pyrazol-3-yl)acetamide
- AZD5991
- [(Z)-16-chloro-11,21,25,61-tetramethyl-11H,21H,61H-10-oxa-4,8-dithia-1(7,3)-indola-2(4,3),6(3,5)-dipyrazola-9(3,1)-naphthalenacyclotridecaphane-12-carboxylic acid]
- CDK
- cyclin-dependent kinase
- CDK5DN
- cyclin-dependent kinase 5 dominant negative
- CI
- combination index
- CP-668863
- N-(3-cyclobutyl-1H-pyrazol-5-yl)-2-(naphthalen-2-yl)acetamide
- Dox
- doxycycline
- GTEx
- genotype-tissue expression
- palbociclib
- 6-acetyl-8-cyclopentyl-5-methyl-2-(5-(piperazin-1-yl)pyridin-2-ylamino)pyrido[2,3-day]pyrimidin-7(8H)-one
- PARP
- poly ADP ribose polymerase
- PDAC
- pancreatic ductal adenocarcinoma
- siRNA
- small interfering RNA
- S63845
- [(R)-2-((5-(3-chloro-2-methyl-4-(2-(4-methylpiperazin-1-yl)ethoxy)phenyl)-6-(5-fluorofuran-2-yl)thieno[2,3-d]pyrimidin-4-yl)oxy)-3-(2-((1-(2,2,2-trifluoroethyl)-1H-pyrazol-5-yl)methoxy)phenyl)propanoic acid]
- TCGA
- The Cancer Genome Atlas
- WEHI-539
- 5-[3-[4-(aminomethyl)phenoxy]propyl]-2-[(8E)-8-(1,3-benzothiazol-2-ylhydrazinylidene)-6,7-dihydro-5H-naphthalen-2-yl]-1,3-thiazole-4-carboxylic acid
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}