


Abstract
The G protein-coupled receptor (GPCR) signaling pathways mediated by trimeric G proteins have been extensively elucidated, but their associated regulatory mechanisms remain unclear. Parathyroid hormone (PTH)/PTH-related protein receptor (PTHR) is a GPCR coupled with Gs and Gq. Gs activates adenylyl cyclases (ACs), which produces cAMP to regulate various cell fates. We previously showed that cell surface expression of PTHR was increased by its direct interaction with a subcortical cytoskeletal protein, 4.1G, whereas PTHR-mediated Gs/AC/cAMP signaling was suppressed by 4.1G through an unknown mechanism in human embryonic kidney (HEK)293 cells. In the present study, we found that AC type 6 (AC6), one of the major ACs activated downstream of PTHR, interacts with 4.1G in HEK293 cells, and the N-terminus of AC6 (AC6-N) directly and selectively binds to the 4.1/ezrin/radixin/moesin (FERM) domain of 4.1G (4.1G-FERM) in vitro. AC6-N was distributed at the plasma membrane, which was disturbed by knockdown of 4.1G. An AC6-N mutant, AC6-N-3A, in which three consecutive arginine residues are mutated to alanine residues, altered both binding to 4.1G-FERM and its plasma membrane distribution in vivo. Further, we overexpressed AC6-N to competitively inhibit the interaction of endogenous AC6 and 4.1G in cells. cAMP production induced by forskolin, an adenylyl cyclase activator, and PTH-(1-34) was enhanced by AC6-N expression and 4.1G-knockdown. In contrast, AC6-N-3A had no impact on forskolin- and PTH-(1-34)-induced cAMP productions. These data provide a novel regulatory mechanism that AC6 activity is suppressed by the direct binding of 4.1G to AC6-N, resulting in attenuation of PTHR-mediated Gs/AC6/cAMP signaling.
Introduction
G protein-coupled receptors (GPCRs) transduce extracellular signals into cells via activation of trimeric G proteins. Among the families of trimeric G proteins, Gs activates adenylyl cyclases (ACs), which results in the production of 3′,5′-cyclic adenosine monophosphate (cAMP). Evidence has accumulated that AC activity and cAMP production are spatially and temporally controlled by various mechanisms including AC-interacting proteins and Gs-coupled GPCR-interacting proteins.
The AC family consists of 10 isoforms; AC1–AC9 are transmembrane ACs, and AC10 is a soluble AC. The former are 12-transmembrane-containing proteins composed of the N-terminus, the first six-transmembrane domain (TM1), the first cytoplasmic loop (C1 loop), the second six-transmembrane domain (TM2) and the second cytoplasmic loop (C2 loop). ACs are activated by Gαs and a direct AC activator, forskolin, and their activities are modulated by Ca2+, calmodulin kinase, protein kinase A, protein kinase C, and regulators of G protein signaling (Cooper and Tabbasum, 2014). Moreover, direct interactions of Gβγ (Brand et al., 2015), A-kinase anchoring proteins (AKAP) 79/150 (Efendiev et al., 2010), and annexin A4, a Ca2+- and phospholipid-binding protein (Heinick et al., 2015) that binds with ACs also regulate AC activities. Recent evidence has demonstrated that cAMP is produced in lipid raft and nonraft regions of the plasma membrane (Agarwal et al., 2014; Averaimo et al., 2016), indicating that both ACs and some of the regulators are functionally distributed in those local regions.
Activation of β2-adrenergic receptor recruits β-arrestins on its carboxyl (C)-terminus, and the recruited β-arrestins further recruit phosphodiesterase 4D, a family of cAMP degradation enzymes, to the receptor. The β2-adrenergic receptor-mediated rate of cAMP production is then decreased by increasing its degradation (Perry et al., 2002). In contrast, activated parathyroid hormone (PTH)/PTH-related protein receptor (PTHR) is internalized by binding β-arrestins with its C-terminus, and the protein complex of PTHR and β-arrestins sustains PTHR-mediated cAMP production on endosome membrane (Ferrandon et al., 2009; Feinstein et al., 2011).
However, further research is necessary to understand the entire scope of the mechanisms that regulate AC/cAMP signaling.
Protein 4.1G is a member of the protein 4.1 family, which interacts with membrane proteins and actin cytoskeleton to provide plasma membrane stability. These proteins are composed of a headpiece (HP), a 4.1-ezrin-radixin-moesin (FERM) domain, a spectrin/actin–binding domain (SABD), and a carboxy-terminal domain (CTD). The HP is a disordered structural domain that interacts with membrane proteins (Nunomura et al., 2011). The FERM domain is an ordered structural domain consisting of three lobes (N-, α- and C-lobes) that also interacts with such membrane proteins as calmodulin, CD44, and band 3 (Nunomura et al., 2011). The SABD is involved in association with actin filament and spectrin. In contrast, the CTD binds to various membrane receptors and increases the cell surface distributions of the receptors, including α7 acetylcholine receptor (Kanno et al., 2013), A1 adenosine receptor (Lu et al., 2004), D2 and D3 dopamine receptors (Binda et al., 2002), and metabotropic glutamate receptors (Shen et al., 2000; Coleman et al., 2003).
PTH plays a key role in maintaining serum calcium and phosphorus levels by affecting bone, kidney, and intestine. We previously showed that PTHR, a Gs- and Gq-coupled GPCR, directly binds to 4.1G through its C-terminus (Saito et al., 2005). 4.1G regulates the plasma membrane distribution of PTHR and potentiation of its Gq/[Ca2+]i signaling (Saito et al., 2005). In contrast, 4.1G attenuates the PTHR/Gs/AC-mediated cAMP production at the plasma membrane without affecting AC6’s plasma membrane distribution (Goto et al., 2013), which is one of the major ACs activated downstream of PTHR (Tovey et al., 2008; Fenton et al., 2014). This is the first case of a plasma membrane-associated cytoskeletal protein suppressing AC activity. However, the molecular mechanism by which 4.1G suppresses PTHR/Gs-mediated AC6 activity remains unclear.
In the present study, we hypothesized that 4.1G directly binds to AC6. We found that direct binding of 4.1G to the N-terminus of AC6 (AC6-N) contributes to the plasma membrane association of AC6-N and the resulting suppression of AC6 cyclase activity. This is responsible for attenuation of PTHR-mediated Gs/AC6/cAMP signaling.
Materials and Methods
Materials and Plasmids.
BL21 Escherichia coli was purchased from Toyobo (Osaka, Japan). Fetal bovine serum (FBS) was purchased from PAA Laboratories GmbH (Pasching, Austria). Dulbecco’s modified Eagle’s medium, mouse anti-DYKDDDDK antibody (cat. no. 012-22384), mouse anti-green fluorescent protein (GFP) antibody (clone mFX73; cat. no. 012-20461), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (cat. no. 016-25523), and forskolin are purchased from Wako (Osaka, Japan). Eagle’s minimum essential medium (EMEM) was obtained from Nissui Pharmaceutical (Tokyo, Japan). Rabbit anti-4.1G antibody (cat. no. PRX-PBR-1004) was purchased from ProteinExpress (Chiba, Japan). pFLAG-MAC vector, mouse anti-FLAG antibody (clone M2; cat. no. F1804), and EDTA-free protease inhibitor cocktail (EDTA-free) were purchased from Sigma-Aldrich (St. Louis, MO). pcDNA3.1(+) vector, Lipofectamine2000, rabbit anti-GFP antibody (cat. no. A6455), protein G Sepharose and Alexa dye-conjugated secondary antibodies were obtained from Life Technologies-Thermo Fisher Scientific (Carlsbad, CA). pMAL-c5X vector and amylose resin beads were purchased from New England BioLabs (Ipswich, MA). pEGFP-C2 vector was obtained from Clontech-TaKaRa (Mountain View, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (cat. no. 7076) and HRP-conjugated anti-rabbit IgG (cat. no. 7074) were purchased from Cell Signaling Technology (Danvers, MA). Rabbit anti-ADCY6 antibody (cat. no. 14616-1-AP) was obtained from Proteintech (Rosemont, IL). Human parathyroid hormone, PTH-(1-34), was obtained from Peptide Institute (Osaka, Japan). AlphaScreen cAMP detection kit was purchased from PerkinElmer (Waltham, MA). A small interfering RNA (siRNA) cocktail (siTrio Full Set) targeting human 4.1G was obtained from B-Bridge International (Mountain View, CA). The cocktail was a mixture of three targets: 5′-GGG AAA GAC GAA AGA GUA ATT-3′, 5′-GGA AAA UGU AGG UGC CCA ATT-3′, and 5′-GGG AAG AUU AAG UAA GAA ATT-3′ (Goto et al., 2013). Control siRNA (Control siRNA-A) was from Santa Cruz Biotechnology (Dallas, TX). Other chemicals were of reagent grade or the highest quality available. pEGFP-C2-AC6-full length (FL) and pcDNA4-DsRed-transmembrane domain (TMD) plasmids were kindly provided by Dr. Colin Taylor (University of Cambridge, UK) (Tovey et al., 2008) and Dr. Shotaro Tanaka (Tokyo Women’s Medical University, Japan) (Tanaka and Takakuwa, 2012), respectively. pME18SFL3-AC3-FL was from National Institute of Technology and Evaluation (Tokyo, Japan). FLAG-tagged full-length of 4.1G (pcDNA3.1(+)-FLAG-4.1G-FL) (Saito et al., 2005) and HA-tagged PTHR (pcDNA3.1(+)-HA-PTHR) (Sugai et al., 2003) were prepared previously.
Plasmid Preparation.
The targeting sequences of human 4.1G-shRNA were synthesized, annealed, and inserted into pCAG-IRES-GFP vector (Li et al., 2011). The sequence of the 4.1G-shRNA was 5′-GGG CAG AGG TTG GGA AAG ACG A-3′.
To clone the intracellular subdomains of AC into pMAL-c5X vector, the nucleotide sequences corresponding with the N-terminus (herein, AC6-N, Met1-Ser151), the C1 loop (AC6-C1, Met351-Pro667), or the C2 loop (AC6-C2, Gln941-Ser1,168) of human AC6 (accession number NM_015270) were amplified by polymerase chain reaction (PCR) from pEGFP-C2-AC6-FL plasmid. The N-terminus of human AC3 (accession number AK122926) (AC3-N, Met1-Leu54) was amplified from pME18SF3-AC3-FL plasmid. Prior to cloning the nucleotide sequences of the human AC7 (accession number NM_001114) (AC7-N, Met1-Pro33) and human AC9 (accession number NM_001116) (AC9-N, Met1-Tyr118) N-termini, total RNA from human embryonic kidney (HEK) 293 cells was reverse-transcribed using specific primers for each gene (AC7; 5′-CGG AGA GTC TTC TCC TGG GCT TG-3′; AC9; 5′-GCA ACT GTG GCG CTT GGA AAG CAC-3′). The nucleotide sequences of AC7-N and AC9-N were then PCR amplified from the respective cDNAs obtained. The amplified PCR products of AC3, AC6, AC7, and AC9 were cloned into pMAL-c5X vector by using NdeI and BamHI.
pcDNA3.1(+)-GFP-AC6-N plasmid was generated by amplifying the nucleotide sequence of GFP-AC6-N using pEGFP-C2-AC6-FL as the template and followed by subcloning into pcDNA3.1(+) using NheI and BamHI.
To obtain the AC6-N-3A mutant sequence, AC6-N was mutagenized by two-step site-directed mutagenesis. First, both Arg29 and Arg30 were mutated into alanine using the primer 5′-GG CAG AAG CGT TCG GCG GCC CGT GGC ACT CGG GC-3′. Arg31 was then mutated to alanine by using the primer 5′-G AAG CGT TCG GCG GCC GCT GGC ACT CGG GCA GGT G-3′. The resulting AC6-N-3A sequence was subcloned into pcDNA3.1(+) vector and pMAL-c5X vector. pMAL-c5X-AC6-N-3K plasmid was obtained by inverse PCR from pMAL-c5X-AC6-N plasmid using primers of 5′-TT CTT CGA ACG CTT CTG CCC ATT GCG TTC-3′ and 5′-G AAG GGC ACT CGG GCA GGT GGC TTC T-3′ (underlined texts indicate mutated nucleotides).
For cloning the nucleotide sequences of 4.1G-HP, -FERM, and -SABD/CTD (accession number NM_001431), the nucleotide sequences corresponding with the amino acid sequences of M1-K221, T215-T510, and Y495-D1005, were PCR amplified using pcDNA3.1(+)-FLAG-4.1G-FL plasmid as the template. The PCR products obtained were cloned into pFLAG-MAC vector using HindIII and KpnI.
The sequences of the resulting plasmids were confirmed by using a genetic analyzer ABI3500 (Applied Biosystems, Waltham, MA).
Cell Culture.
HEK293 cells were obtained from Cell Resource Center for Biomedical Research, Tohoku University, Japan. Mycoplasma contamination in the cells was routinely checked by DAPI staining. HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. The cells were maintained in a humidified atmosphere at 37°C under 5% CO2. Plasmid DNA and/or the siRNA cocktail were transfected into HEK293 cells using Lipofectamine2000 according to the manufacturer’s protocol. The cells were used for experiments 2 days after the transfection. HEK293 cells stably expressing GFP-AC6-FL or HA-PTHR were selected with 1000 μg/ml of G418.
Protein Purification and In Vitro Pull-Down Assay.
Purification of proteins from BL21 E. coli lysate and the in vitro pull-down assay were performed as previously described (Sugai et al., 2003). In brief, E. coli-expressing MPB-fusion proteins or FLAG-fusion proteins were suspended in Tris-buffered saline (TBS; 20 mM Tris-HCl, 150 mM NaCl, pH7.4) supplemented with 1 mM EDTA, 2 mM dithiothreitol, 1× protease inhibitor cocktail, and 1 mM PMSF. The cells were homogenized using a French Pressure Cell Press (FA-078; SLM Aminco). The lysate was centrifuged at 15,000g for 15 minutes at 4°C, and the supernatants were used in the in vitro pull-down assay. The cleared lysates containing MPB-fusion proteins or FLAG-fusion proteins were agitated in buffer A (TBS containing 0.1% NP-40, 1 mM dithiothreitol, and 0.2 mg/ml BSA) for 2 hours at 4°C. After further incubation with amylose resin beads for 1 hour at 4°C, the beads were washed three times with buffer A and once with TBS. The purified protein complexes were solubilized in Laemmli sample buffer (62.5 mM Tris-HCl, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.5 mg/ml bromophenol blue, pH 6.8).
Coimmunoprecipitation Assay.
Sample preparation for the coimmunoprecipitation assay was performed using a slightly modified method from that previously described (Saito et al., 2017). Briefly, HEK293 cells stably expressing GFP-AC6-FL were washed twice with ice-cold TBS and lysed in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% NP-40, 0.5% deoxycholic acid, 0.05% SDS, 1 mM EGTA, and 5% glycerol, pH 7.4) supplemented with 1× protease inhibitor cocktail and 1 mM PMSF. The lysate was passed through a 26-gauge needle more than five times and then centrifuged at 15,000g for 10 minutes at 4°C. The supernatant was precleared by incubation with protein G Sepharose for 30 minutes at 4°C. Equivalent amounts of postnuclear supernatants (on the basis of total protein content) were then incubated with 1.3 μg of mouse anti-GFP antibody (clone mFX73) overnight at 4°C. The equal amount of mouse anti-DYKDDDDK antibody was used as a negative control. The immunoprecipitated proteins were washed three times with lysis buffer and washed once with TBS, and then were solubilized in Laemmli sample buffer.
Protein Expression.
To detect protein expression in HEK293 cells, the cells were washed twice with STE buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EGTA, pH 7.4) and lysed with STE buffer supplemented with 1% Trition X-100, 1× protease inhibitor cocktail, and 1 mM PMSF. The lysate was centrifuged at 15,000g for 10 minutes at 4°C, and equivalent protein (on the basis of total protein content) in the supernatant obtained was mixed with 5× Laemmli sample buffer.
Coomassie Brilliant Blue Staining.
Protein samples separated on the SDS-PAGE were visualized by Coomassie Brilliant Blue (CBB) staining and detected by an Odyssey Infrared Imager (LI-COR, Lincoln, NE).
Western Blotting.
Protein samples were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membrane. The membrane was blocked with 5% nonfat dry milk in TBS containing 0.1% Tween 20 for 1 hour at room temperature. The blot was probed with mouse anti-DYKDDDDK antibody (1:2000), rabbit anti-4.1G antibody (1:2000), rabbit anti-GFP antibody (1:2000), or mouse anti-GAPDH antibody (1:5000) overnight at 4°C. The blot was further probed with the appropriate HRP-conjugated secondary antibodies for 1 hour at room temperature. Signals were obtained by incubating the blots with chemiluminescent HRP substrate and digitally recorded by ChemiDoc XRS+/Image Laboratory software (Bio-Rad, Hercules, CA). Band intensities were quantified by the software.
Immunofluorescence.
HEK293 cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline and their immunofluorescences were performed as previously described (Saito et al., 2017, 2018). The following antibodies were used: rabbit anti-GFP (1:1000), mouse anti-FLAG (clone M2, 1:1000), fluorescence-conjugated secondary antibodies (1:400). Confocal microscope images were acquired with a 63× objective on a confocal microscope LSM 780 (Zeiss, Oberkochen, Germany). The cells expressing DsRed-TMD were fixed with 4% PFA and then observed by confocal microscopy without the immunostaining process. Fluorescence profiles were plotted by Zen (Zeiss). For quantification of GFP-AC6 proteins at the plasma membrane, regions of interest (ROI) at the DsRed-TMD-positive plasma membrane were selected in cells chosen at random, and GFP fluorescence was quantified using ImageJ. The fluorescence intensity at the ROI was calculated as a percentage of the intensity of the whole cell.
AlphaScreen cAMP Assay.
Sample preparation for the cAMP assay was performed as previously described with some modifications (Goto et al., 2013). HEK293 cells cultured in poly-l-lysine-coated 48-well plates were rinsed once in prewarmed EMEM buffered with 20 mM HEPES, pH 7.4 (herein, EMEM-HEPES), and then preincubated in the buffer for 30 minutes at 37°C. Cells were then treated with 10 μM forskolin or 0.3–30 nM PTH-(1-34) in the presence of 100 μM 3-isobutyl-1-methylxanthine (IBMX), a phosphodiesterase inhibitor, for 10–14 minutes at 37°C. The reaction was terminated by a quick rinse in phosphate-buffered saline and the subsequent replacement of the buffer with 150 μl of 2.5% perchloric acid. After a 20-minute incubation at room temperature, 100 μl of the supernatant was neutralized by adding 11 μl of 4.2 N KOH, and the precipitated potassium perchlorate was separated by centrifugation. The cAMP concentration in the supernatant was measured using an AlphaScreen cAMP detection kit and a microplate reader PHERAStar (BMG Labtech, Ortenberg, Germany) according to the manufacturer’s protocol. The cAMP concentration was normalized to the total protein concentration.
Statistics.
Graph-drawing and statistical analysis were performed with Prism v8.0 (GraphPad Software). The numbers of experiments and samples were specified before any data were obtained. Data are presented as the means ± S.D. or S.E.M. as indicated in figure legends. One-way analysis of variance followed by Tukey’s test (as a post-hoc test) or Student’s t test was performed to determine statistically significant differences. A P value <0.05 was considered significant. The P values analyzed are reported in figures.
Results
AC6 Interacts with 4.1G In Vivo.
We previously showed that 4.1G plays a key role in suppressing AC activity (Goto et al., 2013), and AC6 is one of the major ACs functionally activated downstream of PTHR (Tovey et al., 2008). On the basis of these results, we investigated whether 4.1G interacts with AC6 in intact cells. First, to investigate the intracellular distributions of both proteins, we established HEK293 cells stably expressing GFP-fused full-length AC6 (GFP-AC6-FL/HEK293), transiently expressed FLAG-tagged full-length 4.1G (FLAG-4.1G-FL), and measured the immunofluorescence of both proteins by confocal microscopy. We found that GFP-AC6-FL and FLAG-4.1G-FL were colocalized at the cell periphery (Fig. 1, A and B), even though the majority of GFP-AC6-FL was distributed at an intracellular organelle (possibly at the endoplasmic reticulum) (Fig. 1A). Expression of both GFP-AC6-FL and FLAG-4.1G-FL were confirmed by Western blotting (Supplemental Fig. 1B). Next, the interaction of AC6 with 4.1G was examined by a coimmunoprecipitation assay using the GFP-AC6-FL/HEK293 cells. The anti-GFP antibody immunoprecipitated GFP-AC6-FL and also endogenous 4.1G (Fig. 1C, lane 4). In contrast, GFP-AC6-FL was not immunoprecipitated by an anti-DYKDDDDK antibody from GFP-AC6-FL/HEK293 cells, which was used as the negative control (Fig. 1C, lane 3) nor by anti-GFP antibody using parental HEK293 cells (Fig. 1C, lane 2). These results suggest that AC6 physically interacts with 4.1G in vivo.

AC6 interacts with 4.1G in vivo. (A) Intracellular distribution of GFP-AC6-FL and FLAG-4.1G-FL. HEK293 cells stably expressing GFP-AC6-FL were transiently expressed with FLAG-4.1G-FL. The cells were colabeled with rabbit anti-GFP antibody (green) and mouse anti-FLAG antibody (clone M2, red). The boxed area indicates the enlarged region. One of the representative 10 images is shown. Scale bar, 10 μm. (B) Fluorescence intensity (F.I.) profile plots from (a) to (a’) shown in (A). Arrowheads indicate the position of the cell periphery. (C) Interaction of GFP-AC6-FL and endogenous 4.1G in HEK293 cells. Parental HEK293 cells or HEK293 cells stably expressing GFP-AC6-FL (GFP-AC6-FL/HEK293) were subjected to a coimmunoprecipitation assay. Mouse anti-GFP antibody (mFX73) or mouse anti-DYKDDDDK antibody as a negative control was used for immunoprecipitation. Both immunoprecipitants (IP) and whole lysates were immunoblotted (IB) with rabbit anti-4.1G or rabbit anti-GFP antibody. Representative immunoblots of five independent experiments are shown.
The N-Terminus of AC6 Directly Binds to the FERM Domain of 4.1G In Vitro.
To investigate the direct binding of 4.1G to AC6, we purified recombinant AC6 and 4.1G proteins from E. coli. Three intracellular regions of AC6—the N-terminus, the C1 loop, and the C2 loop—were fused with maltose-binding protein (MBP; 43 kDa) to produce MBP-AC6-N (59 kDa), MBP-AC6-C1 (82 kDa), and MBP-AC6-C2 (68 kDa), respectively (Fig. 2A). The amino acid sequences of these regions are shown in Supplemental Fig. 2. The HP, FERM domain, and SABD/CTD of 4.1G were FLAG-tagged to produce FLAG-4.1G-HP (26 kDa), FLAG-4.1G-FERM (36 kDa), and FLAG-4.1G-SABD/CTD (58 kDa), respectively (Fig. 2B). The results of the in vitro pull-down assay showed that MBP-AC6-N specifically bound to FLAG-4.1G-FERM (Fig. 2C, upper). FLAG-4.1G-HP, FLAG-4.1G-FERM, and FLAG-4.1G-SABD/CTD proteins expressed in E. coli lysates were separated by SDS-PAGE and stained with CBB, suggesting that equal amounts of FLAG-4.1G-FERM proteins were applied in the experiments (Supplemental Fig. 1A). The apparent molecular weight of FLAG-4.1G-HP in the SDS-PAGE gel was much higher than its theoretical molecular weight with unspecified causes. To confirm that equal amounts of protein were loaded in each lane, MBP-AC6 proteins pulled down with amylose resin beads were visualized by CBB staining (Fig. 2C, lower).

The N-terminus of AC6 directly binds to the FERM domain of 4.1G. (A) Schematic of the domain structure of AC6. AC6 consists of five domains: the N-terminus (N), the first six-transmembrane domain (TM1), the first cytoplasmic loop (C1), the second six-transmembrane domain (TM2), and the second cytoplasmic loop (C2). Intracellular subdomains of AC6 were fused with maltose binding protein (MBP) to produce MBP-AC6-N, MBP-AC6-C1, and MBP-AC6-C2, respectively. (B) Schematic of the domain structure of 4.1G. 4.1G is composed of four major domains: the headpiece (HP), the 4.1-ezrin-moesin-radixin domain (FERM), the spectrin-actin binding domain (SABD), and the carboxy-terminal domain (CTD). Subdomains of 4.1G were FLAG-tagged to produce FLAG-4.1G-HP, FLAG-4.1G-FERM, and FLAG-4.1G-SABD/CTD, respectively. (C) In vitro pull-down assay. MBP or MBP-AC6 proteins were incubated with FLAG-4.1G proteins and further incubated with amylose resin beads. Proteins bound to the amylose resin were separated by SDS-PAGE and detected by immunoblotting (IB) by using mouse anti-DYKDDDDK antibody (upper). MBP or MBP-AC6 proteins purified by amylose resin beads were stained with CBB (lower). (-): MBP (43 kDa); N: MBP-AC6-N (59 kDa); C1: MBP-AC6-C1 (82 kDa); and C2: MBP-AC6-C2 (68 kDa). Arrowheads indicate the apparent molecular weight of FLAG-4.1G-HP (26 kDa), -FERM (36 kDa), and -SABD/CTD (58 kDa). Representative immunoblots of three independent experiments are shown.
AC6-N Associates with the Plasma Membrane through Its Interaction with 4.1G.
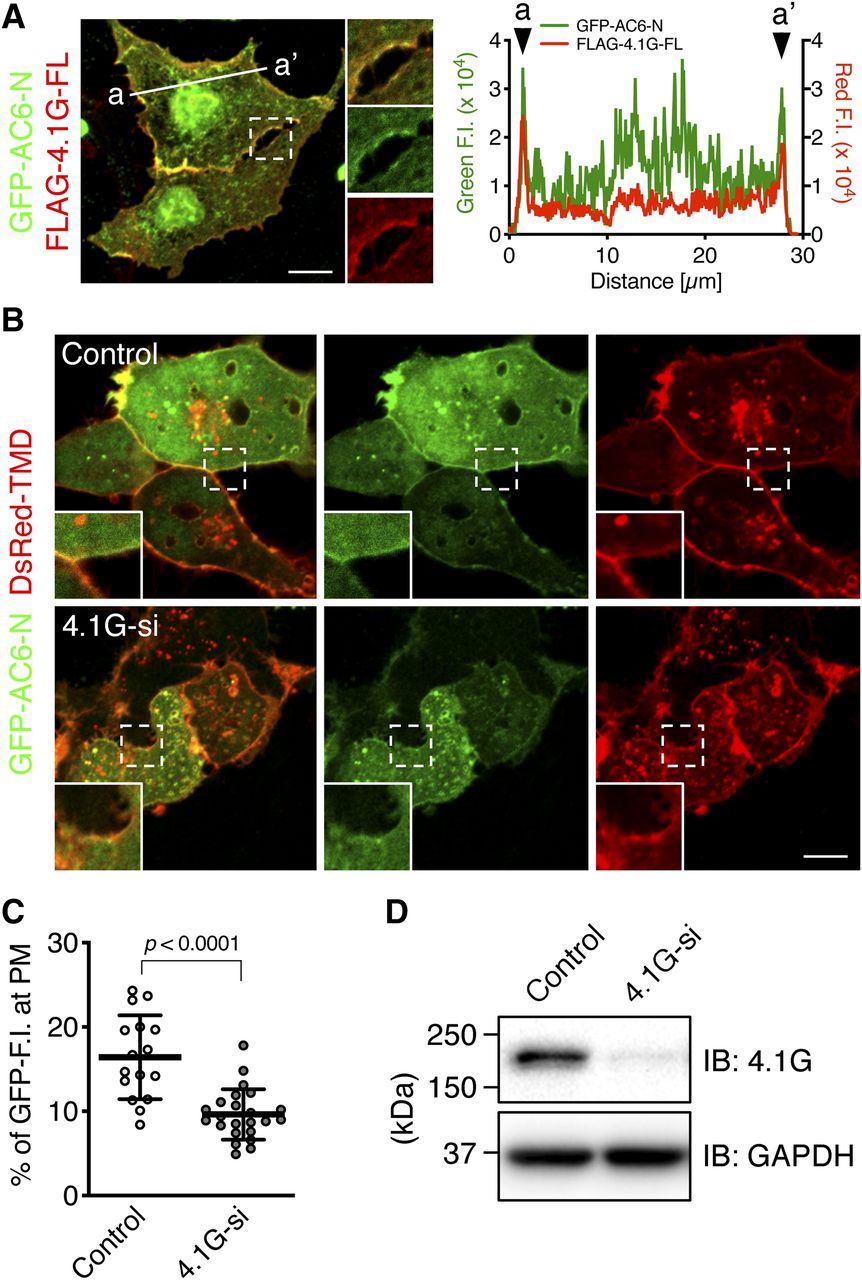
We next hypothesized that AC6-N, one of the intracellular subdomains of AC6, distributes at the plasma membrane through its binding to 4.1G. To answer this question, we examined the intracellular distributions of GFP-AC6-N and FLAG-4.1G-FL in HEK293 cells by immunofluorescence confocal microscopy. First, we found that GFP-AC6-N was codistributed with FLAG-4.1G-FL at the cell periphery (Fig. 3, A and B), and it was distributed at the plasma membrane and the cytoplasm (Fig. 3C, Control), as did a previous study (Thangavel et al., 2009). We next investigated whether the plasma membrane distribution of GFP-AC6-N depended on the presence of 4.1G. Endogenous 4.1G was knocked down by transfection of an siRNA cocktail, and the plasma membrane was visualized by exogenously expressed DsRed-TMD (Tanaka and Takakuwa, 2012). The result showed that the localization of GFP-AC6-N at the plasma membrane was reduced by 4.1G-knockdown (Fig. 3C, 4.1G-si). To quantify the plasma membrane distribution of GFP-AC6-N, the plasma membrane ROI was selected by monitoring the DsRed-TMD signal at the cell periphery, and the fluorescence intensity of GFP-AC6-N at the ROI was expressed as a percentage of the whole-cell fluorescence intensity. The quantification indicated that 4.1G-knockdown reduced the presence of GFP-AC6-N at the plasma membrane (41% reduction) (Fig. 3D). Both the molecular weight of GFP-AC6-N (45 kDa) (Supplemental Fig. 1C) and knockdown efficiency of endogenous 4.1G (Fig. 3E) were confirmed by Western blotting. In contrast to GFP-AC6-N, the distribution of GFP-AC6-FL at the plasma membrane was not altered by 4.1G-knockdown (98% expression vs. Control) (Fig. 4), consistent with our previous study (Goto et al., 2013). These results collectively demonstrate that 4.1G controls the association of AC6-N with the plasma membrane without affecting the distribution of AC6-FL at the plasma membrane.

AC6-N is associated with the plasma membrane via 4.1G. (A) Intracellular distribution of GFP-AC6-N and FLAG-4.1G-FL. HEK293 cells transiently expressed with GFP-AC6-N and FLAG-4.1G-FL were labeled with rabbit anti-GFP (green) and mouse anti-FLAG (clone M2, red) antibodies. The boxed areas indicate the enlarged regions. One of the representative 16 images is shown. Scale bar, 10 μm. (B) Fluorescence intensity (F.I.) profile plots from (a) to (a’) shown in (A). Arrowheads indicate the position of the cell periphery. (C) Plasma membrane distribution of GPF-AC6-N. HEK293 cells expressing both GPF-AC6-N and DsRed-TMD were also transfected with (lower) or without (upper) 4.1G-siRNA (4.1G-si). The cells fixed with 4% PFA were observed by confocal microscopy without immunolabeling. One representative image from each transfection is shown. The boxed areas indicate the enlarged regions. Scale bar, 10 μm. (D) Quantification of the fluorescence intensity of GFP-AC6-N distributed at DsRed-TMD-labeled plasma membrane (PM) region in (C). n = 16 (control) or 23 (4.1G-si). Data are presented as the means ± S.D. Student’s t test was performed for statistical analysis. (E) Knockdown efficiency of 4.1G-si in HEK293 cells. The cell lysates were immunoblotted (IB) with rabbit anti-4.1G or mouse anti-GAPDH antibody. Representative immunoblots of three independent experiments are shown.

4.1G does not affect the plasma membrane distribution of GPF-AC6-FL. (A) Intracellular distribution of GFP-AC6-FL. HEK293 cells transiently expressed with both GFP-AC6-FL and DsRed-TMD were further transfected with (lower) or without (upper) of 4.1G-siRNA (4.1G-si). The cells fixed with 4% PFA were observed by confocal microscopy without immunolabeling. One representative image from each transfection is shown. The boxed areas indicate the enlarged regions. Scale bar, 10 μm. (B) Quantification of the fluorescence intensity of GFP-AC6-FL distributed at DsRed-TMD-labeled plasma membrane (PM) region in (A). n = 22 (control) or 30 (4.1G-si). Data are presented as the means ± S.D. Student’s t test was performed for statistical analysis.
A Three Consecutive Arginine Sequence in AC6-N Is a Binding Site to 4.1G-FERM.
We sought to identify the sequence in AC6-N responsible for its binding to 4.1G-FERM. A previous study demonstrated that three consecutive, positively charged amino acids are responsible for binding to an ezrin/radixin/moesin domain domain (Yonemura et al., 1998). AC6-N contains three consecutive arginine residues (Arg29-Arg31; herein, R29-RR31) (Fig. 5A; Supplemental Fig. 2A), which we mutated to alanine residues (herein, A29-AA31); this construct was named AC6-N-3A (Fig. 5A). We first determined that the mutations affected the apparent binding affinity (KD) of MBP-AC6-N to FLAG-4.1G-FERM by an in vitro pull-down assay (Supplemental Fig. 3). Next, we used the KD concentration (212 nM) of MBP-AC6-N and MBP-AC6-3A in the assay and found that the binding of MBP-AC6-N-3A was markedly lower than that of MBP-AC6-N (82% ± 4.6% reduction) (Fig. 5B). These results indicate that AC6-N directly binds with 4.1G-FERM through its R29-RR31 sequence.

The three consecutive arginine sequence in the N-terminus of AC6 is essential for binding to the FERM domain of 4.1G. (A) Amino acid sequences of AC6-Ns. Triple-arginine and triple-alanine sequences in AC6-N and AC6-N-3A are underlined. Amino acid residues are numbered above the sequences. (B) In vitro pull-down assay of MBP-AC6-Ns and FLAG-4.1G-FERM. FLAG-4.1G-FERM that bound to MBP-AC6-Ns (212 nM) were visualized by SDS-PAGE and immunoblotting (IB) using mouse anti-DYKDDDDK antibody (upper). FLAG-4.1G-FERM proteins contained in the lysates were immunoblotted with anti-DYKDDDDK antibody (middle). MBP and MBP-AC6-Ns pulled down by amylose resin beads were separated by SDS-PAGE and visualized by CBB staining (lower). MBP (-) (43 kDa), MBP-AC6-N (59 kDa), MBP-AC6-N-3A (59 kDa), and FLAG-4.1G-FERM (36 kDa). Representative immunoblots of three independent experiments are shown. (C) Plasma membrane distribution of GPF-AC6-N or -3A. HEK293 cells were expressed with GPF-AC6-N or -3A in the presence of DsRed-TMD. The cells fixed with 4% PFA were observed by confocal microscopy without immunolabeling. One representative image from each transfection is shown. The boxed areas indicate the enlarged regions. Scale bar, 10 μm. (D) Quantification of the fluorescence intensities of GFP-AC6-N or -3A distributed at DsRed-TMD-labeled plasma membrane (PM) region in (C). n = 26 (WT) or 26 (3A). Data are presented as the means ± S.D. Student’s t test was performed for statistical analysis.
On the basis of our result that the plasma membrane distribution of AC6-N was decreased by 4.1G-knockdown and the binding of AC6-N-3A was markedly lower than that of AC6-N, we speculated that GFP-AC6-N-3A was weakly (or not) associated with the plasma membrane. We then examined the distribution of GFP-AC6-N-3A in HEK293 cells. GFP-AC6-N was predominantly distributed at the DsRed-TMD-labeled plasma membrane (Fig. 5C, AC6-N), in contrast, less GFP-AC6-N-3A signal was observed at the plasma membrane (Fig. 5C, AC6-N-3A). The quantification and analysis of these results indicated that the amount of GFP-AC6-N-3A at the plasma membrane was reduced by 22% compared with that of GFP-AC6-N (Fig. 5D). GFP-AC6-N-3A also weakly codistributed with FLAG-4.1G-FL at the cell periphery (Supplemental Fig. 4). The molecular weight of GFP-AC6-N-3A (45 kDa) was confirmed by Western blotting (Supplemental Fig. 1C). These results demonstrate that the binding of the R29-RR31 sequence of AC6-N to 4.1G-FERM is required for its association with the plasma membrane.
To investigate contribution of the positive charges in the three consecutive amino acids to binding to 4.1G-FERM, we substituted the R29-RR31 sequence of AC6-N with lysine residues (herein, K29-KK31); this construct was named AC6-N-3K (Supplemental Fig. 5A). As a result of in vitro pull-down assay, we found that MBP-AC6-N and MBP-AC6-N-3K were equally bound to FLAG-4.1G-FERM (binding of MBP-AC6-N-3K was 96% ± 2.7% of that of MBP-AC6-N) (Supplemental Fig. 5B), suggesting that three consecutive positive charges play an important role in binding to 4.1G-FERM.
Here, we tested whether three consecutive arginine and/or lysine residues are the only consensus sequences that can bind to 4.1G-FERM. For this purpose, among AC1, AC3, AC6, AC7, and AC9 that are endogenously expressed in HEK293 cells (Goto et al., 2013), AC3, AC7, and AC9 were chosen. Note that their N-termini do not contain three consecutive arginine and/or lysine residues (Supplemental Fig. 6A). The N-termini of AC3, AC7, and AC9 were fused with MBP, and their bindings to FLAG-4.1G-FERM were examined by an in vitro pull-down assay. Compared with MBP-AC6-N, we found that MBP-AC9-N equally bound and MBP-AC3-N very weakly bound to FLAG-4.1G-FERM, whereas MBP-AC7-N did not bind at all (Supplemental Fig. 6B, upper). The equal amount of FLAG-4.1G-FERM protein was applied in each reaction (Supplemental Fig. 6B, middle), and the comparable amounts of MBP-proteins were pulled down by amylose resin beads (Supplemental Fig. 6B, lower). Interestingly, AC9-N possesses a highly positively charged cluster, R74-RQKK78, rather than a consecutive sequence of positively charged residues (Supplemental Fig. 6A), suggesting that three consecutive arginine and/or lysine residues are not the only requisite for binding to the FERM domain. In contrast, AC6-C1, which has an K558-RK560 sequence, did not bind to 4.1G-FERM (Fig. 2C; Supplemental Fig. 2B).
The Interaction of 4.1G to AC6-N Attenuates AC6 Cyclase Activity.
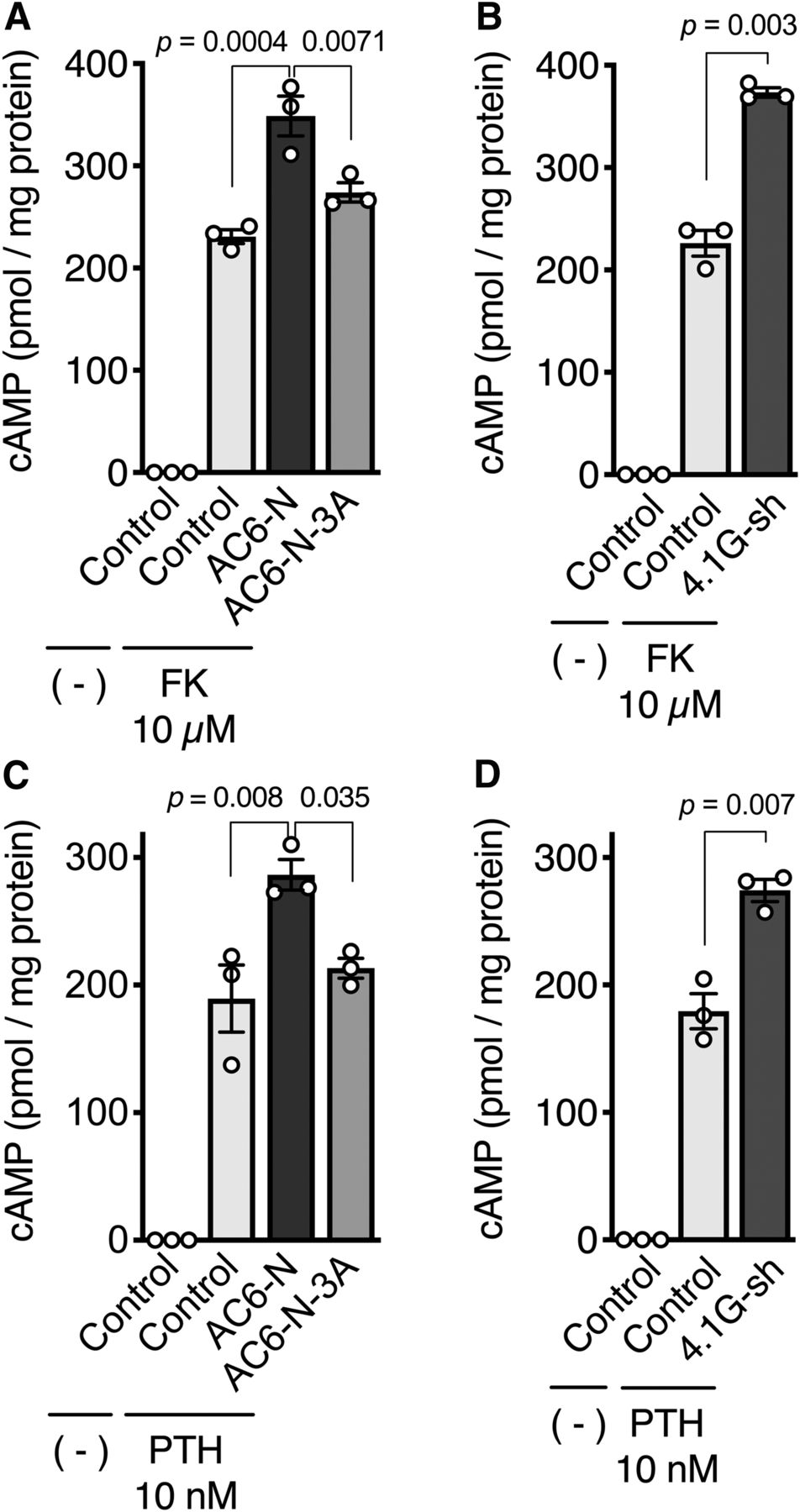
To investigate the physiologic impact of direct binding between 4.1G and AC6 on cyclase activity, GFP-AC6-N was overexpressed to inhibit the interaction of endogenous AC6 and 4.1G in HEK293 cells. When the cells were treated with forskolin, a direct activator of adenylyl cyclases, cAMP production increased in control cells and was further enhanced by overexpression of GFP-AC6-N, as well as shRNA-mediated knockdown of 4.1G (herein, 4.1G-sh) (Fig. 6, A and B). In contrast, GFP-AC6-N-3A did not alter forskolin-induced cAMP production (Fig. 6A). In our assay system, cAMP production was undetectable in the absence of forskolin. Like our previous observation (Goto et al., 2013), we ascertained that overexpression of GFP-AC6-Ns or knockdown of 4.1G did not alter the amount of endogenous AC6 protein expressed in HEK293 cells (Fig. 7). These data suggest that the direct interaction of 4.1G to AC6-N suppresses AC6 activity.

The association of AC6-N with the plasma membrane is critical for AC6 activity. (A and B) Effect of (A) overexpression of GFP-AC6-Ns or (B) knockdown of 4.1G in forskolin-induced cAMP production. HEK293 cells were exogenously expressed with (A) GFP-AC6-N or -3A or (B) transfected with 4.1G-shRNA (4.1G-sh). The cells were treated with 10 μM forskolin (FK) in the presence of 100 μM IBMX for 10 minutes at 37°C. (C and D) Effect of (C) overexpression of GFP-AC6-Ns or (D) knockdown of 4.1G in PTH-(1-34)-induced cAMP production. HEK293 cells stably expressing HA-PTHR were transiently expressed with (C) GFP-AC6-N or -3A or (D) transfected with 4.1G-sh. The cells were treated with 10 nM PTH-(1-34) in the presence of 100 μM IBMX for 14 minutes at 37°C. The intracellular cAMP concentration was analyzed by AlphaScreen cAMP assay as described in Materials and Methods. (A and C) Data are presented as the means ± S.E.M., n = 3; one-way analysis of variance followed by Tukey’s tests were performed within the drug-treated cells. (B and D) Data are presented as the means ± S.E.M., n = 3, Student’s t tests were performed between the drug-treated cells.

The expression of endogenous AC6 protein is not altered by AC6-N-expression or 4.1G-knockdown. (A) Expression of endogenous AC6 protein in HEK293 cells transiently expressing GFP, GFP-AC6-N, or GFP-AC6-N-3A was detected by Western blotting. The samples were immunoblotted (IB) with rabbit anti-AC6 antibody, rabbit anti-GFP antibody, or mouse anti-GAPDH antibody. (B) Quantification of (A). Data are presented as the means ± S.D. from three independent experiments. (C) Expression of endogenous AC6 protein in HEK293 cells transfected with 4.1G siRNA cocktail. The samples were immunoblotted (IB) with rabbit anti-AC6 antibody, rabbit anti-4.1G antibody, or mouse anti-GAPDH antibody. (D) Quantification of (C). Data are presented as the means ± S.D. from two independent experiments.
To gain further insight into the effects of GFP-AC6-N- or GFP-AC6-N-3A-overexpression on PTHR-mediated cAMP production, we first established an HEK293 cell line stably expressing HA-PTHR. When the cells were treated with various doses of PTH-(1-34), cAMP production was increased in a dose-dependent manner with a maximum response at 10 nM (Supplemental Fig. 7). Next, we treated the cells with 10 nM PTH-(1-34) to evaluate the effects of GFP-AC6-N-overexpression and 4.1G-knockdown. We found that GFP-AC6-N overexpression and 4.1G-knockdown, but not GFP-AC6-N-3A overexpression, enhanced cAMP production compared with that in control cells (Fig. 6, C and D). These results suggest that the interaction of 4.1G with AC6 is responsible for the attenuation of PTHR-mediated Gs/AC6/cAMP signaling.
Discussion
We previously showed that suppression of AC activity by 4.1G is responsible for attenuation of PTHR/Gs/AC-mediated cAMP production (Goto et al., 2013). The present study describes a novel observation that 4.1G directly interacts to AC6-N. The direct binding plays a key role in plasma membrane distribution of AC6-N, and the resulting suppression of AC6 cyclase activity. On the basis of these observations, we developed a theoretical model in which AC6-N associates with the plasma membrane through binding to 4.1G, resulting in low AC6 activity (Fig. 8A). In contrast, the dissociation of AC6-N from 4.1G and the plasma membrane results in high AC6 activity (Fig. 8B).

Theoretical models. (A) In normal condition, the three consecutive arginine sequence (RRR) in the N-terminus of AC6 is associated with the plasma membrane via 4.1G, and AC6 activity is restricted. (B) When the N-terminus of AC6 is dissociated from the plasma membrane (e.g., lower expression level of 4.1G or overexpression of AC6-N), AC6 activity is high.
To be more precise, our theoretical model is supported by the evidence described following: 1) Plasma membrane association of AC6-N was disturbed by 4.1G-knockdown, 2) the forskolin-induced cAMP production was enhanced by overexpression of an AC6-N fragment, which presumably inhibited the binding of AC6 and 4.1G, 3) the R29-RR31 sequence in AC6-N was required for its binding to 4.1G-FERM and association of AC6-N with the plasma membrane, 4) overexpression of AC6-N-3A mutant had little effect on the forskolin-induced cAMP production, and 5) a smaller amount of AC6-N-3A distributed at the plasma membrane. However, the correlation between plasma membrane association of AC6 and its cyclase activity is the missing piece of the puzzle. Our theoretical model would be clarified by unraveling the structure-activity correlation of AC6.
4.1G plays a key role in plasma membrane association of AC6-N. The present study also suggests a possibility that 4.1G is not the only molecule necessary for the plasma membrane association of AC6-N. We showed that the amount of GFP-AC6-N at the plasma membrane was decreased by 41% by 4.1G-knockdown and a lesser amount of GFP-AC6-N-3A distributed at the plasma membrane than of GFP-AC6-N (22% reduction). On the other hand, 4.1G expression was nearly abolished by 4.1G siRNA, and binding of MBP-AC6-N-3A to FLAG-4.1G-FERM was only 18% of that of MBP-AC6-N. This evidence suggests that other molecules also contribute to the association of AC6-N with the plasma membrane. We also found that plasma membrane distribution of GFP-4.1G-FL was not altered by 4.1G-knockdown, suggesting that regions other than AC6-N play pivotal roles in the distribution of AC6-FL at the plasma membrane.
How does the direct binding of 4.1G to AC6 suppress AC6 activity? Gαs proteins activate all types of transmembrane ACs through GPCRs (Willoughby and Cooper, 2007). When the ACs are activated by Gαs or forskolin, the C1 and C2 loops dimerize to stimulate cyclase activity (Sunahara et al., 1997; Tesmer et al., 1997). Because our results herein show that 4.1G does not bind to either loops, one of the possible mechanisms is that 4.1G coordinately regulates AC6 activity through other AC6-N-binding proteins, such as Gβγ and AKAP79/150. Gβγ associates with the plasma membrane through farnesylation or geranylgeranylation of the Gγ subunit (Wedegaertner et al., 1995) and activates AC5 and AC6 through binding to their N-termini and C1/C2 loops (Brand et al., 2015). It is expected that 4.1G hinders the interaction between Gβγ and AC6-N, and the dissociation may prevent the dimerization of C1 and C2 loops of AC6. In contrast, AKAP79/150 can associate with the N-termini of AC5 and AC6 and suppress their activities (Efendiev et al., 2010). AKAP79 contains three polybasic regions required for its targeting to the plasma membrane (Dell’Acqua et al., 1998; Delint-Ramirez et al., 2011). Thus, we deduce that 4.1G also associates with the polybasic regions of AKAP79. The complex of AC6-4.1G-AKAP79 would contribute to the suppression of AC6 activity. It was recently considered that functional-protein complexes (e.g., signalosomes) transduce adaptive cellular signaling (Ellisdon and Halls, 2016). It is of interest to fully elucidate how the activities of AC6 and other AC types are regulated by such protein complexes.
It was unclear whether suppression of Gs/AC6/cAMP signaling by 4.1G was specific to PTHR or not (Goto et al., 2013). The present study shows that 4.1G reduces the maximum cAMP production mediated through PTHR. Because the reduction results from the suppression of AC6 activity, 4.1G would suppress Gs/AC6/cAMP signaling of various GPCRs. In other words, 4.1G would be a global regulator of Gs/AC6/cAMP signaling.
We previously showed that 4.1G directly interacted with the C-terminus of PTHR through its CTD (Saito et al., 2005), which led to the prediction that 4.1G concurrently forms a functional complex with both PTHR and AC6 (PTHR-4.1G-AC6). We are, however, dismissive of the formation of the complex for two reasons: First, the increase in cell surface expression of PTHR by 4.1G results in augmentation of its Gq/[Ca2+]i signaling (Saito et al., 2005), and it conflicts with the suppression of AC6 signaling by 4.1G shown in the present study. Second, it is conceivable that PTHR and AC6 are in distinct locations on the plasma membrane. PTHR mainly distributes in nonlipid raft regions (Tovey and Taylor, 2013), whereas AC6 is associated with lipid rafts or caveolae (Ostrom et al., 2000). 4.1G would distribute in both lipid rafts and nonlipid rafts, as does 4.1R in the red cell membrane (Salzer and Prohaska, 2001). Collectively, it is considered that 4.1G forms separate protein complexes with PTHR and AC6. The binding of 4.1G with AC6, and lacking formation of a protein complex of PTHR-4.1G-AC6, would be an efficient way to suppress PTHR-mediated Gs/AC6/cAMP signaling.
Whole-cell expression and/or cell surface distribution of PTHR can be varied depending on the cell environment, such as agonist stimuli. PTHR is internalized by agonist stimulation (Ferrari et al., 1999; Sugai et al., 2003), and internalization of PTHR is involved in sustained cAMP production on endosome membranes (Ferrandon et al., 2009; Feinstein et al., 2011). The present study is, however, limited to the functional and physical interactions of AC6 and 4.1G on the plasma membrane. Thus, it is valuable to consider whether changes in PTHR levels alter the interaction of AC6 and 4.1G by performing stoichiometric analyses. Moreover, to uncover whether 4.1G predominantly regulates cAMP production on the plasma membrane, we are interested in elucidating the role of 4.1G in the internalization of PTHR and cAMP production on the endosome membrane.
In the present study, we also showed that 4.1G selectively binds to AC6-N and AC9-N. These results demonstrate that three, consecutive, positively charged amino acids, as found in the R29-RR31 sequence of AC6-N, are not the only the consensus sequence for binding to the FERM domain. AC9-N does not contain three, consecutive, positively charged amino acids but contains R74-RQKK78, suggesting that clusters of positively charged amino acids are required. An R114-RQR117 sequence in AC9-N would have weaker binding to 4.1G-FERM because of its similarity to AC3-N (K71-RQR74), which weakly binds to 4.1G-FERM. In contrast, it appears that amino acid residues proximal to the cluster would also affect binding to 4.1G-FERM. Although AC6-C1 includes a K558-RK560 sequence, it did not bind to 4.1G-FERM. The presence of two glutamate residues adjacent to the K558-RK560 sequence (K558-RKEE562) provides two predictions: 1) The negative charges of the glutamate residues electrically negate the positive charges of the K558-RK560 sequence, or 2) access of 4.1G-FERM to AC6-C1 is hindered by the negative charges, because negative charges in the 4.1G-FERM would be required for its binding to the clusters of positively charged amino acids. For the binding to FERM domain, these findings collectively imply that: 1) the cluster should be composed of more than three amino acids, 2) the percentage of positively charged amino acids in the cluster should be more than 80%, and 3) a negatively charged amino acid(s) should not be located proximal to the cluster. These prerequisites for binding to FERM domains should be scrutinized in the future. It would also be interesting to elucidate whether AC9 activity is suppressed by the direct binding of 4.1G in the future.
The function of 4.1G on AC6-mediated cAMP production would depend on the level of 4.1G expression. For example, 4.1G is expressed in the neonatal kidney (Walensky et al., 1998) and embryonic kidney cells as shown in the present study, but not in an adult kidney (Ramez et al., 2003). PTHR/Gs/AC6/cAMP signaling contributes to the excretion of phosphorus in the kidney cells (Fenton et al., 2014), implying that 4.1G suppresses the excretion predominantly in the neonatal stage. A paucity of PTH action, including low PTH production in hypoparathyroidism, leads to low phosphorus excretion and high serum phosphorus (e.g., hyperphosphatemia). It is of interest to investigate whether 4.1G is applied to a therapeutic target in neonatal hyperphosphatemia.
In conclusion, we have revealed that 4.1G, a plasma membrane-associated cytoskeletal protein, directly binds to the N-terminus of AC6, resulting in suppression of AC6 activity. A cluster of arginine residues, R29-RR31, in the N-terminus of AC6 is responsible for the association with the plasma membrane by binding to the FERM domain of 4.1G. The present study provides new insights into the diversity of regulatory mechanisms for GPCR signaling.
Acknowledgments
We greatly appreciate Dr. Colin Taylar (University of Cambridge, UK), Dr. Shotaro Tanaka (Tokyo Women’s Medical University, Japan), and National Institute of Technology and Evaluation for providing pEGFP-C2-AC6-FL, pcDNA4-DsRed-TDM and pME18SF3-AC3-FL-WT plasmids, respectively. We would also like to acknowledge the Biomedical Research Core (Tohoku University Graduate School of Medicine) and Sayuri Saito for technical support.
Authorship Contributions
Participated in research design: Saito, Yanagisawa, Sukegawa.
Conducted experiments: Saito, Cui, Hirano, Li.
Contributed new reagents or analytic tools: Saito, Sato, Sukegawa.
Performed data analysis: Saito, Cui, Sato, Sukegawa.
Wrote or contributed to the writing of the manuscript: Saito, Sato, Sukegawa.
Footnotes
- Received February 22, 2019.
- Accepted July 30, 2019.
This work was supported by the Grant-in-Aid for Scientific Research from the Japan Society for Promotion of Science (No. 15K20856 and No. 18K06125 to M.S., No. 24590311 to T.Y., No. 18K06885 to T.S., and No. 21200007, No. 21590267, No. 26460331 to J.S.), Takeda Science Foundation, Nishinomiya Basic Research Fund (to M.S.), and Shokei Gakuin University (to J.S.). Conflict of interest: The authors declare that they have no conflict of interest.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AC
- adenylyl cyclase
- AKAP
- A-kinase anchoring proteins
- cAMP
- 3′,5′-cyclic adenosine monophosphate
- CBB
- Coomassie Brilliant Blue
- CTD
- carboxy-terminal domain
- C-terminus
- carboxyl terminus
- EMEM
- Eagle’s minimum essential medium
- FERM
- 4.1/ezrin/radixin/moesin
- FL
- full-length
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- HEK
- human embryonic kidney
- HP
- headpiece
- HRP
- horseradish peroxidase
- IBMX
- 3-isobutyl-1-methylxanthine, 1-methyl-3-(2-methylpropyl)-7H-purine-2,6-dione
- MBP
- maltose-binding protein
- PCR
- polymerase chain reaction
- PFA
- paraformaldehyde
- PTH
- parathyroid hormone
- PTHR
- PTH/PTH-related protein receptor
- PMSF
- phenylmethylsulfonyl fluoride
- ROI
- regions of interest
- SABD
- spectrin/actin–binding domain
- shRNA
- short hairpin RNA
- siRNA
- small-interfering RNA
- TBS
- Tris-buffered saline
- TMD
- transmembrane domain
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}