


Abstract
The constitutive androstane receptor (CAR) plays an important role in xenobiotic metabolism, energy homeostasis, and cell proliferation. Antagonism of the CAR represents a key strategy for studying its function and may have potential clinical applications. However, specific human CAR (hCAR) antagonists are limited and conflicting data on the activity of these compounds have been reported. 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide (PK11195), a typical peripheral benzodiazepine receptor ligand, has been established as a potent hCAR deactivator in immortalized cells; whether it inhibits hCAR activity under physiologically relevant conditions remains unclear. Here, we investigated the effects of PK11195 on hCAR in metabolically competent human primary hepatocytes (HPH) and HepaRG cells. We show that although PK11195 antagonizes hCAR in HepG2 cells, it induces the expression of CYP2B6 and CYP3A4, targets of hCAR and the pregnane X receptor (PXR), in HPH, HepaRG, and PXR-knockout HepaRG cells. Utilizing a HPH-HepG2 coculture model, we demonstrate that inclusion of HPH converts PK11195 from an antagonist to an agonist of hCAR, and such conversion was attenuated by potent CYP3A4 inhibitor ketoconazole. Metabolically, we show that the N-desmethyl metabolite is responsible for PK11195-mediated hCAR activation by facilitating hCAR interaction with coactivators and enhancing hCAR nuclear translocation in HPHs. Structure-activity analysis revealed that N-demethylation alters the interaction of PK11195 with the binding pocket of hCAR to favor activation. Together, these results indicate that removal of a methyl group switches PK11195 from a potent antagonist of hCAR to an agonist in HPH and highlights the importance of physiologically relevant metabolism when attempting to define the biologic action of small molecules.
Introduction
The constitutive androstane receptor (CAR) is a xenobiotic sensor primarily expressed in the liver that regulates expression of genes associated with drug-metabolizing enzymes and transporters (Zelko and Negishi, 2000; Qatanani and Moore, 2005; Omiecinski et al., 2011). Under physiologic conditions, CAR is sequestered in the cytoplasm of hepatocytes and translocates to the nucleus upon activation, in which it dimerizes with the retinoid X receptor, recruits coactivators such as SRC-1, and induces the transcription of its target genes (Kawamoto et al., 1999; Pascussi et al., 2007). Activation of CAR leads to the coordinated induction of a cellular defensive network in the liver, which enables hepatocytes to metabolize and excrete foreign compounds. However, induction of important drug-metabolizing enzymes through CAR activation can also significantly alter the levels of toxic metabolites and lead to unexpected drug-drug interactions (Tolson and Wang, 2010; Kobayashi et al., 2015). Recently, novel functions of CAR beyond modulation of drug disposition have come to light, including its roles in energy metabolism and cell proliferation (Yamamoto et al., 2004; Dong et al., 2009; Gao et al., 2009). Thus, understanding the molecular basis of CAR modulators would not only benefit early prediction of drug-drug interactions but also offer potential therapeutic choices for metabolic disorders and cancers.
Although CAR is sequestered in the cytoplasm and must translocate to the nucleus to transactivate gene expression in primary hepatocytes and intact livers, it is spontaneously localized in the nucleus and exhibits constitutive activity in immortalized cell lines (Kawamoto et al., 1999; Li et al., 2009). This unique feature makes the investigation of chemical-mediated CAR modulation rather challenging. We have previously identified 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide (PK11195), a peripheral benzodiazepine receptor ligand, as a potent and selective antagonist of human CAR (hCAR) with negligible effects on its rodent counterparts (Li et al., 2008). At low micromolar concentrations, PK11195 effectively represses hCAR-mediated gene expression in cell lines and has been used as a research tool for studying the molecular mechanisms of hCAR activation (Anderson et al., 2011; Lynch et al., 2015). Notably, although a potent antagonist of hCAR, PK11195 also activates the human pregnane X receptor (PXR) efficiently (Li et al., 2008; Anderson et al., 2011). Given that CAR and PXR share an overlapping array of target genes (Moore et al., 2000) it is challenging to delineate the exact effects of PK11195 on CAR and PXR in cells that express both receptors. To this end, the fact that PK11195 induces the expression of CYP2B6, the primary target of hCAR, and CYP3A4, the primary target of human (hPXR), in human primary hepatocytes (HPHs) has been attributed to its activation of hPXR. Nevertheless, unlike the prototypical hPXR activator rifampicin, which preferentially induces the expression of CYP3A4 over CYP2B6, PK11195 favorably enhances the expression of CYP2B6 over CYP3A4, challenging this assumption.
It is well established that CAR plays a key role in determining xenobiotic metabolism and disposition through transactivation of numerous genes in the liver. However, whether and how hepatic metabolism can influence CAR action is not adequately understood. A previous study showed that although omeprazole represents a prototypical activator of the aryl hydrocarbon receptor, its degradation metabolite omeprazole-sulfide is a pure antagonist of the aryl hydrocarbon receptor (Gerbal-Chaloin et al., 2006). In screening of antimalarials and their major metabolites for CAR activation, Burk et al. (2012) found that artemisinin derivatives and metabolites differentially affect activities of CAR. Clearly, the metabolic capacity of a cellular system can be a key determinant for the biologic function of a given compound including its role in nuclear receptor activation. Thus, we hypothesize that PK11195 induction of CYP2B6 and CYP3A4 in HPHs is not fully mediated by the activation of PXR, instead, PK11195 is converted from an antagonist to an agonist of hCAR in the metabolically competent HPH.
In the current study, we demonstrate that PK11195 is N-demethylated to a metabolite that robustly activates CAR in HPHs. Using a PXR-knockout (KO) HepaRG cell line, we confirm PK11195 can induce the expression of CYP2B6/CYP3A4 independent of PXR. A coculture system that adds metabolism to CAR luciferase reporter assays was used to determine how metabolism influences the agonist/antagonist feature of PK11195. Mammalian two-hybrid assays and molecular modeling were used to elucidate the molecular basis and structural features of PK11195 and its metabolites in the modulation of CAR activity.
Materials and Methods
Chemicals and Biologic Reagents.
Phenobarbital (PB), rifampicin (RIF), ketoconazole (KET), and PK11195 were obtained from Sigma-Aldrich (St. Louis, MO). 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime (CITCO) was obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). (R)-N-Desmethyl PK11195 (ND-PK) was obtained from ABX Advanced Biochemical Compounds (Radeberg, Germany). 1-(2-Chlorophenyl)isoquinoline-3-carboxylic acid (COOH-PK) was obtained from Biogene Organics (The Woodlands, TX). Polymerase chain reaction (PCR) primers were synthesized by Integrated DNA Technologies (Coralville, IA). HepaRG wild-type and PXR-KO cells and the cell culture medium were obtained from Sigma-Aldrich. Optima LC/MS Grade water (H2O), acetonitrile (ACN), and formic acid were purchased from Fisher Scientific (Pittsburg, PA). All chemicals and reagents were used without further purification.
Culture and Treatment of HPHs and HepaRG Cells.
HPHs were isolated using a modified two-step perfusion protocol (LeCluyse et al., 2005) from human liver specimens obtained from the University of Maryland Medical Center with prior approval by the Institutional Review Board at the University of Maryland at Baltimore or obtained from Bioreclamation In Vitro Technologies (Baltimore, MD). The age and sex of each liver donor used are detailed in Table 1. Hepatocytes with viability over 90% were seeded at 0.75 × 106 cells/well in 12-well collagen-coated plates as described previously (Faucette et al., 2006). After attachment at 37°C in a humidified atmosphere of 5% CO2, hepatocytes were cultured in serum-free William’s E Medium supplemented with insulin, transferrin, and selenium, 0.1 μM dexamethasone, 100 U/ml penicillin, and 100 μg/ml streptomycin, and overlaid with Matrigel (0.25 mg/ml). Thirty-six hours after seeding, HPHs were treated with vehicle control (0.1% [dimethylsulfoxide (DMSO)], CITCO (1 μM), RIF (10 μM), PB (1 mM), PK11195 (10 μM), ND-PK (10 μM), or COOH-PK (10 μM) for 24 or 72 hours before harvesting cells to collect RNA or protein, respectively. In separate experiments, wild-type and PXR-KO HepaRG cells were plated in 12-well plates (1 × 105 cells/well) and cultured for 21 days following Sigma-Aldrich’s instructions to induce differentiation before treatment with compounds as described previously.
Age and sex of liver donors
Real-Time Reverse-Transcription PCR.
Total RNA was isolated from cells using TRIzol reagent (ThermoFisher, Rockford, IL) and reverse transcribed using a High Capacity cDNA archive kit (Applied Biosystems, Foster, CA) according to the manufacturer’s instructions. Real-time PCR assay was performed using SYBR Green PCR Mastermix (Qiagen, Germantown, MD) on an ABI StepOnePlus real-time PCR system (Applied Biosystems). He primer sequences for CYP2B6, CYP3A4, and glyceraldehyde-3-phosphate dehydrogenase are as follows: CYP2B6, 5′-AGACGCCTTCAATCCTGACC-3′ and 5′-CCTTCACCAAGACAAATCCGC-3′; CYP3A4, 5′-GTGGGGCTTTTATGATGGTCA-3′ and 5′-GCCTCAGATTTCTCACCAACACA-3′; and glyceraldehyde-3-phosphate dehydrogenase, 5′-CCCATCACCATCTTCCAGGAG-3′ and 5′-GTTGTCATGGATGACCTTGGC-3′. Expression values were quantified using the following equation: fold over control = 2ΔΔCt method, where ΔCt represents the differences in cycle threshold numbers between the target gene and glyceraldehyde-3-phosphate dehydrogenase, and ΔΔCt represents the relative change in these differences between control and treatment groups.
Western Blot Analysis.
Protein samples extracted from treated cells were electrophoretically separated on SDS-PAGE gels (4%–12%) and transferred to polyvinylidine fluoride membranes. Subsequently, membranes were incubated with primary antibodies against CYP2B6 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), CYP3A4 (1:5000; Sigma-Aldrich), or β-actin (1:50,000, Sigma-Aldrich) at 4°C overnight. Blots were developed with West Pico chemiluminescent substrates (ThermoFisher) after incubation with horse-radish peroxidase secondary antibodies.
HepG2 Cell Transfection and HepG2/HPH Coculture.
HepG2 or HepG2-CAR-CYP2B6 stable cells as described previously (Lynch et al., 2015) were cultured in 24-well plates (1 × 105 cells/well) at 37°C and 5% CO2 for 24 hours. HepG2 cells were cotransfected with CYP2B6-2.2k reporter (60 ng/well), hCAR1+A expression vector (30 ng/well), and pRL-TK (10 ng/well) by using X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich) following the manufacturer’s instruction. Twenty-four hours after transfection, cells were treated with solvent (0.1% DMSO) or test compounds at indicated concentrations. Subsequently, cell lysates were assayed for firefly activities normalized against the activities of Renilla using a luciferase kit (Promega, Madison, WI). In separate experiments, HPHs were plated on collagen-coated cover slips with the corners bent upward in a 24-well plate. After preincubation of vehicle control and test compounds with HPHs for 4 hours, both the media and HPH-containing cover slips were transferred to new 24-well plates containing transfected HepG2 cells or the HepG2-CAR-2B6 stable line and incubated for 24 hours before measurement of luciferase activities. Data from a representative liver donor are shown in Fig. 3B-C and represent the mean ± S.D. of three individual transfections.
Liquid Chromatography (LC)–Mass Spectrometry (MS) Measurement of PK11195 Metabolism.
Complete William’s E Medium containing 10 µM PK11195 was added to HPHs cultured in 24-well plates 36 hours after seeding. Media (450 µl) were collected from each well at 0 and 1 hours and frozen immediately at −80°C. Samples were thawed on ice before adding 50 µl of medium to 450 µl of ice-cold methanol/H2O (8:1, v/v). After centrifugation at 16,000g for 30 minutes, 200 µl of supernatant was transferred to a separate tube and dried down before resuspension in 200 µl H2O/ACN (1:1, v/v) with 0.1% formic acid. Authentic standards were prepared to 1 µM in H2O/ACN (1:1, v/v) with 0.1% formic acid. LC-MS/MS analysis was performed on a TSQ Quantum Ultra Triple Stage Quadrupole Mass Spectrometer coupled to an Ultimate 3000 RS Liquid Chromatogram System (Thermo Scientific, Waltham, MA). The LC separation was performed on a Waters (Milford, MA) BEH C18 column (2.1 × 50 mm, 1.7 µm) operated at 30°C. Solvents A and B consisted of 0.1% formic acid in H2O and 0.1% formic acid in ACN, respectively. The gradient program was 0.0–0.5 minutes, 50% B; 0.5–2.0 minutes, gradient to 95% B; 2.0–3.5 minutes, 95% B; 3.5–4.0 minutes, gradient to 50% B; and 4.0–5.0 minutes, 50% B. The flow rate was 0.5 ml/min and injection volume was 5 µl. Tandem mass spectrometry was performed in the positive-ion mode and the electrospray ionization source parameters were as follows: spray voltage, 3000; capillary temperature, 325; sheath gas pressure, 40; auxiliary gas pressure, 15; capillary offset, 35; and tube lens offset, 80. Selected reaction monitoring was used for mass detection with the following transitions: PK11195 (m/z 353.1 → 238.0), ND-PK (m/z 339.1 → 238.0), and COOH-PK (m/z 284.0 → 238.0). Data collection and analysis were performed using Xcalibur V 2.1 (Thermo Scientific).
Mammalian Two-Hybrid Assay.
COS1 cells seeded in 24-well plates were transfected with 110 ng of the reporter gene plasmid pG5luc, 80 ng of expression plasmids encoding the respective VP16-AD/hCAR fusions, 40 ng of expression plasmids encoding GAL4-DBD/coregulatory fusions, and 20 ng of reference plasmid pRL-TK, each well using X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich). Twenty-four hours after transfection, the cells were treated with solvent (0.1% DMSO), CITCO (1 µM), PK11195 (10 µM), or ND-PK (10 µM) for 24 hours. Luciferase activities were measured in cell lysates using the Dual-Luciferase Kit (Promega). Data represent the mean ± S.D. of three individual transfections.
Nuclear Translocation of CAR in HPHs.
HPHs were plated in collagen-coated 24-well plates and infected with adenovirus-expressing enhanced yellow fluorescent protein-tagged hCAR (EYFP-hCAR) as described previously (Li et al., 2009). Twenty-four hours after infection, HPHs were treated with vehicle control (0.1% DMSO), PB (1 mM), PK11195 (10 µM), ND-PK (10 µM), or COOH-PK (10 µM) for another 8 hours. After treatment, cells were fixed with 4% paraformaldehyde, stained with 1 µg/ml 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) for 30 minutes, and EYFP-hCAR localization in hepatocytes was visualized on a Nikon Eclipse TI fluorescent microscope (Nikon, Melville, NY). Quantitative distribution of EYFP-hCAR was analyzed using General Analysis in the Nikon Elements AR High Content Analysis software package (version 4.50.00). Nuclear localization was defined and quantified as the percentage of total enhanced yellow fluorescent protein that overlaps with 4′,6-diamidino-2-phenylindole. Data from a representative liver donor are shown in Fig. 7A-B and represent the mean ± S.D. of five individual images for each treatment.
Molecular Modeling.
The hCAR/ligand-binding domain protein crystal structure (Protein Data Bank identification number 1XVP) was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org). The PK11195 and ND-PK molecular structures were generated and obtained from ChemAxon Chemicalize (http://chemicalize.com) and the CITCO and CAR inhibitor not PXR activator 1 (CINPA1) structures were obtained from National Center for Biotechnology Information PubChem (http://pubchem.ncbi.nlm.nih.gov/). Discovery Studio (version 4.5.0.15071; Biovia, San Diego, CA) was used to remove water and ligands from the crystallographic data and isolate the D chain protein that contains the crystal structure of CAR, which was subsequently protonated at pH 7.0. A binding site was defined based on the CITCO binding cavity and defined as an 11.5 Å radius sphere at 24.972 (x), 54.702 (y), and 29.512 (z). The receptor-ligand docking was performed in Discovery Studio using the CDOCKER protocol (Wu et al., 2003). Briefly, the docking parameters were set to generate 255 conformations for each ligand and return the top 10 docks with the lowest CDOCKER energy, which is the sum of the receptor-ligand interaction energy and internal ligand strain. The complete parameters can be found in Supplemental Fig. 1 and Supplemental Table 1. The ligand-receptor interactions of the docked molecules were analyzed and visualized in Discovery Studio.
Statistical Analysis.
All data are expressed as the mean ± S.D. Statistical comparisons were made using one-way analysis of variance with Dunnett’s post-test or two-way analysis of variance with Bonferroni post-test as needed. Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
PK11195 Induces the Expression of CYP2B6 and CYP3A4 in HPHs.
We first examined the effects of PK11195, a known hCAR antagonist, on the expression of CYP2B6 and CYP3A4, two prototypical targets for hCAR and hPXR, in HPHs prepared from liver donors 107 and 122. As shown in Fig. 1, PB, CITCO, RIF, and PK11195 at selected concentrations robustly induced CYP2B6 and CYP3A4 at mRNA and protein levels in both liver donors. As expected, CITCO (a selective activator of hCAR) and RIF (a selective activator of hPXR) preferentially induced the expression of CYP2B6 and CYP3A4, respectively. Intriguingly, PK11195 at 10 µM, a concentration at which it represses the activity of hCAR by more than 85% in HepG2 cells (Li et al., 2008), markedly induced the expression of both CYP2B6 and CYP3A4 without a clear preference, mimicking PB, a dual activator of hCAR and hPXR. These findings call into question whether PK11195 is as an antagonist of hCAR in physiologically relevant cells and challenge the assumption that PK11195 induction of cytochrome P450 (P450) in HPHs relies on PXR activation.

PK11195 induces CYP2B6 and CYP3A4 expression in HPHs. Primary hepatocytes from liver donors 107 (A and B) and 122 (C and D) were treated with 1 mM PB, 1 µM CITCO, 10 µM RIF, or 10 µM PK11195 for 24 or 72 hours to analyze mRNA or protein expression, respectively. Results are expressed as fold over control, mean ± S.D. (n = 3); ***P < 0.001.
Induction of CYP2B6 and CYP3A4 by PK11195 in PXR-KO HepaRG Cells.
HepaRG cells have been validated as a promising surrogate for HPHs, and importantly, fully differentiated HepaRG cells exhibit proper CAR cellular localization and maintain physiologically relevant metabolic capacity, which are not present in most immortalized cell models (Jackson et al., 2016). The PXR-KO HepaRG cell line obtained from Sigma-Aldrich is a newly generated cell line that does not express functional PXR (Williamson et al., 2016). As expected, PK11195 and other known CAR/PXR modulators induced the expression of CYP2B6 and CYP3A4 mRNA and protein in wild-type HepaRG cells in a trend that mirrors what was observed in HPHs (Fig. 2, A and B). Notably, in PXR-KO HepaRG cells PK11195 significantly induced both CYP2B6 and 3A4 expression at mRNA and protein levels, although induction of CYP2B6 and CYP3A4 by RIF was fully abrogated (Fig. 2, C and D). These data suggest that differential metabolism of PK11195 in the physiologically relevant HPH/HepaRG cells versus the immortalized HepG2 cells may contribute to the observed PXR-independent induction of CYP2B6 and CYP3A4.
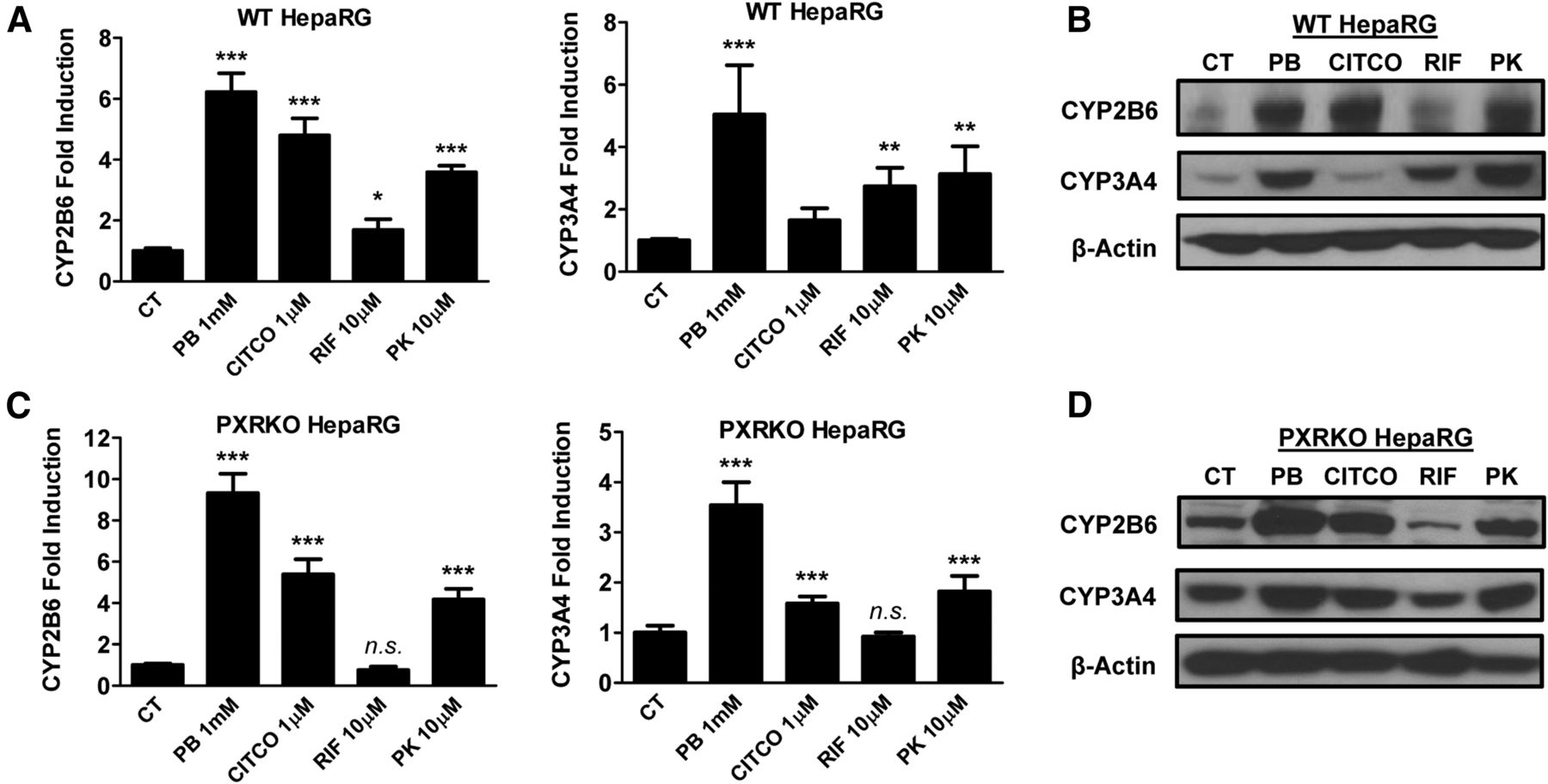
PK11195 induces CYP2B6 and CYP3A4 expression in HepaRG cells independent of PXR. Wild-type and PXR-KO HepaRG cells were cultured for 21 days in accordance with Sigma-Aldrich instructions to induce differentiation. Differentiated HepaRG cells were treated with 1 mM PB, 1 µM CITCO, 10 µM RIF, or 10 µM PK11195 for 24 or 72 hours to analyze mRNA (A and C) or protein expression (B and D), respectively. Data represent the mean ± S.D. (n = 3); n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
PK11195 Is Metabolized to a hCAR Activator in HPHs.
Lack of metabolism is a major limitation of almost all studies using immortalized cell lines, including HepG2 cells. A HPH-HepG2 coculture model was established as depicted in Fig. 3A, which introduces the metabolism-competent HPHs into the culture environment shared with HepG2 cells. In agreement with previous reports, PK11195 concentration dependently inhibits the constitutive hCAR activity in HepG2 cells without the presence of HPHs (Fig. 3B, open bars). Results from the coculture with HPHs from a representative liver donor indicate that PK11195 significantly increased the luciferase activity of the CYP2B6 reporter in the presence of hCAR (Fig. 3B, solid bars) or its low-basal alternative hCAR1+A (Fig. 3C). These findings suggest that PK11195 is converted from a CAR antagonist to an agonist in the presence of HPHs. To further explore the contribution of metabolism in this antagonism/agonism conversion, KET, a potent inhibitor of CYP3A4, the most abundant hepatic P450 isoform in humans, was cotreated with different concentrations of PK11195 in HepG2-CAR-2B6 cells with and without HPH coculture. Repression of CAR activity by PK11195 was not influenced by the presence of KET in HepG2-CAR-2B6 cells alone (Fig. 3D). On the other hand, when HPHs were cocultured with HepG2-CAR-2B6 cells, KET significantly inhibited the conversion of PK11195 (1 and 10 µM) from a CAR antagonist to a CAR activator (Fig. 3E). Taken together, these results validate the use of the coculture system to investigate the mechanistic effects of metabolism on CAR activity and suggest that CYP3A4 contributes to the biotransformation of PK11195 in HPHs.

PK11195 is metabolized to a CAR activator in HepG2-HPH coculture. HPHs were plated on plastic cover slips with the corners bent upward and incubated with test compounds for 4 hours before the media and the cover slips were transferred to a 24-well plate containing HepG2 cells and incubated for 24 hours (A). HepG2-CAR-2B6 stable line (B) or transiently transfected with CAR1+A and CYP2B6-2.2K (C) were treated with PK11195 and CITCO at indicated concentrations with or without coculture with HPHs for 24 hours. HepG2-CAR-2B6 cells alone (D) or cocultured with HPHs (E) were treated with PK11195 alone or cotreated with 8 µM KET for 24 hours. Luciferase activity was measured as described in Materials and Methods. Data represent the mean ± S.D. (n = 3); *P < 0.05; ***P < 0.001.
PK11195 Is Metabolized to ND-PK and COOH-PK in HPHs.
Previous studies postulated that PK11195 is metabolized into two major metabolites, ND-PK and COOH-PK, which are formed through N-demethylation and amide hydrolysis, respectively (Fig. 4A) (Roivainen et al., 2009). To confirm that these metabolites were generated in our experimental system, PK11195 (10 µM) was incubated with HPHs and media was collected at 0 and 1 hours. These samples were prepared and analyzed by LC-MS/MS as detailed in Materials and Methods to determine whether the proposed ND-PK and COOH-PK metabolites of PK11195 were formed in HPHs. The 1 µM standard mix exhibited chromatographic separation and reproducible retention times of 1.81, 0.76, and 2.14 for PK11195, COOH-PK, and ND-PK, respectively (Fig. 4B). As expected, PK11195 was abundantly present in HPH culture medium at 0 hour, while the amounts of COOH-PK and ND-PK were negligible (Fig. 4C). However, the peaks for COOH-PK and ND-PK significantly increased in intensity after 1 hour in HPH culture, indicating that PK11195 is metabolized by HPHs to COOH-PK and ND-PK (Fig. 4D). To our knowledge, this is the first report confirming that the COOH-PK and ND-PK metabolites of PK11195 are generated by HPHs, and these metabolites require further study to determine whether they mediate CAR activation.

PK11195 is metabolized to COOH-PK and ND-PK in HPHs. Proposed PK11195 metabolism pathway (A). An authentic 1 µM standard mix was run on LC-MS/MS as described in Materials and Methods and retention times for PK11195, COOH-PK, and ND-PK were determined to be 1.81, 0.76, and 2.14, respectively (B). Media containing 10 µM PK11195 was cultured with HPHs and harvested at 0 hour (C) and 1 hour (D) to determine whether PK11195 is metabolized by HPHs to the metabolites of interest.
The (R)-N-Desmethyl Metabolite of PK11195 Mediates hCAR Activation.
After confirming that COOH-PK and ND-PK were generated in our experimental system, we tested these metabolites for hCAR activation in HepG2-based luciferase assays. As shown in Fig. 5A, ND-PK activated hCAR in a concentration-dependent manner while COOH-PK had no effect on CAR activity. ND-PK activation of hCAR was further confirmed in the CAR1+A/CYP2B6-2.2k reporter assay, where ND-PK robustly activates hCAR to a level similar to the positive control (CITCO), while COOH-PK exhibits negligible CAR activation (Fig. 5B). In separate experiments, the mammalian two-hybrid assay revealed that PK11195 could significantly repress the interaction between hCAR and SRC-1 or GRIP1, while ND-PK moderately enhanced hCAR recruitment of these coactivators in a manner similar to that by CITCO (Fig. 5, C and D). Notably, ND-PK markedly rescued PK11195-mediated repression of SRC-1/CAR interaction, while only minimally affecting PK11195-repressed binding of GRIP1 to hCAR. Furthermore, ND-PK significantly induced CYP2B6 and CYP3A4 mRNA and protein expression in the PXR-KO HepaRG cells, indicating that ND-PK can induce P450 expression independent of PXR (Fig. 5, E and F). Overall, these results identify ND-PK as the metabolite of PK11195 that is responsible for PK11195-mediated CAR activation in metabolically competent hepatic cells.

The (R)-N-desmethyl (ND) metabolite of PK11195 mediates CAR activation. HepG2-CAR-2B6 cells (A) and HepG2 cells transfected with CAR1+A and CYP2B6-2.2k (B) were treated with vehicle control (CT), ND-PK, COOH PK, or CITCO at indicated concentrations for 24 hours before luciferase activities were determined. Mammalian two-hybrid assays were performed in COS1 cells measuring interaction between CAR/SRC-1 (C) or CAR/GRIP1 (D) as detailed in Materials and Methods. Transfected cells were treated with CITCO, PK11195, ND-PK, or ND-PK+PK1195 for 24 hours before determining luciferase activity. PXR-KO HepaRG cells were treated with vehicle control, PB (1 mM), CITCO (1 µM), RIF (10 µM), ND-PK (10 µM), or COOH-PK (10 µM) and harvested for mRNA (E) or protein (F) expression analysis. Data represent the mean ± S.D. (n = 3); n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
ND-PK Induces CYP2B6, CYP3A4 Expression, and Nuclear Translocation of hCAR in HPHs.
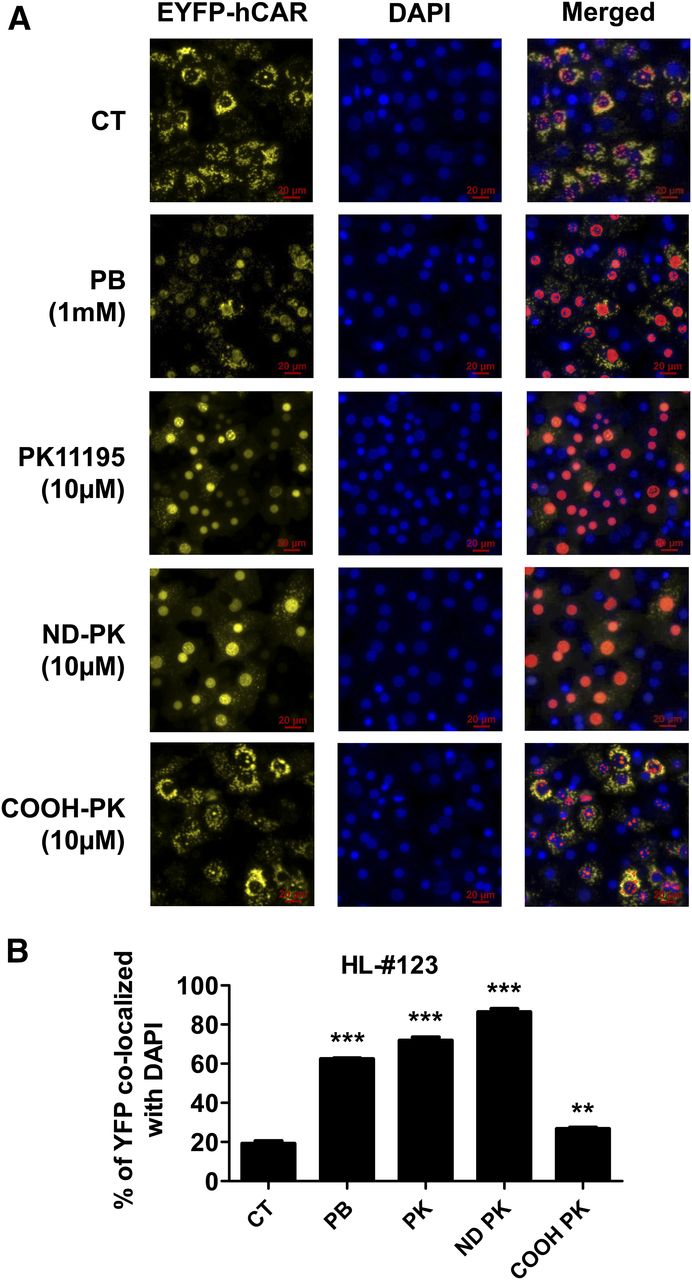
To fully characterize the effect of ND-PK on hCAR activation and target gene induction, HPHs from two liver donors (120 and 121) were treated with multiple concentrations of PK11195, ND-PK, and COOH-PK. Our results indicate that ND-PK concentration dependently and potently induced CYP2B6 and CYP3A4 at both mRNA and protein levels, while COOH-PK only exhibited negligible effects on the expression of these genes (Fig. 6, A–D). Nuclear translocation of CAR in HPHs has been regarded as the essential step in its activation. To test the nuclear translocation of CAR by PK11195 metabolites, HPHs were infected with adenovirus-expressing EYFP-hCAR overnight before treatment with compounds for 8 hours. Without activation, EYFP-hCAR was predominantly expressed in the cytoplasm of HPHs, and as expected the prototypical CAR activator PB as well as PK11195 induced significant hCAR nuclear accumulation (Fig. 7, A and B). Importantly, ND-PK at 10 µM robustly increased nuclear translocation of hCAR, while COOH-PK (10 µM) caused negligible CAR nuclear accumulation, which correlate well with their capacity for P450 induction. These data further support that ND-PK is the metabolite of PK11195 that activates hCAR in physiologically relevant systems.

ND-PK induces CAR target genes in HPHs. Primary hepatocytes from liver donors 120 (A and B) or 121 (C and D) were treated with PB (1 mM), CITCO (1 µM), RIF (10 µM), or increasing concentrations (0.1, 1, and 10 µM) of PK11195, ND-PK, or COOH-PK for 24 or 72 hours to analyze mRNA (A and C) or protein (B and D) expression, respectively. Data represent the mean ± S.D. (n = 3).

ND-PK induces CAR nuclear translocation in HPHs. Primary hepatocytes from liver donor 123 were infected with adenovirus-expressing EYFP-hCAR for 24 hours and treated with 1 mM PB, 10 µM PK11195, 10 µM ND-PK, or 10 µM COOH-PK for 8 hours before being fixed with 4% paraformaldehyde, stained with 1 µg/ml 4′,6-diamidino-2-phenylindole (DAPI), and visualized on a Nikon Eclipse TI fluorescent microscope. Quantitation of nuclear localization was determined using General Analysis in the Nikon Elements AR High Content Analysis software package and defined as the percentage of enhanced yellow fluorescent protein (YFP) that overlaps with DAPI. Representative images (A) and quantitative analysis (B) of nuclear localization are shown. Data represent the mean ± S.D. of five individual images; **P < 0.01; ***P < 0.001.
Computational Modeling of PK11195 and ND-PK.
The metabolism of a potent CAR antagonist (PK11195) and its conversion into a potent CAR activator (ND-PK) with a difference of only one methyl group provided a unique opportunity to probe the structure-activity relationship for CAR through molecular modeling. Docking studies used the 1XVP (http://www.rcsb.org) crystal structure of CITCO bound to the CAR ligand-binding domain to identify PK11195 and ND-PK interactions with different residues in the binding pocket (Xu et al., 2004). Upon validation of our model by docking CITCO into the ligand-binding domain, both PK11195 and ND-PK were docked and found to interact with many of the same residues in the binding pocket, suggesting that they bind in similar conformations (Fig. 8A). However, PK11195 interacts with residues important to CAR activity such as V199, Y224, and Y326, including the N-methyl group interacting with H203, whereas the demethylated form of PK11195 (ND-PK) does not (Fig. 8, B and C); notably, PK11195 and the CAR antagonist CINPA1 (Fig. 8D) share these interactions, which may contribute to their antagonism of hCAR (Cherian et al., 2016). The demethylation of PK11195 allows it move away and not interact with these important residues in the CAR binding pocket, which may explain how the difference of one methyl group between PK11195 and ND-PK can have such a substantial effect on CAR activity. These docking studies determined the structure-activity relationship of PK11195 and ND-PK with CAR and demonstrated that even small alterations in ligand structure, such as the demethylation of PK11195, have the potential to alter its interactions with amino acids in the CAR binding pocket and lead to large differences in CAR activity.

N-Demethylation of PK11195 leads to reduced interaction with residues important to CAR activity. The hCAR/ligand-binding domain crystal structure (1XVP) was prepared in Biovia Discovery Studio and docking was carried out using the CDOCKER protocol as detailed in Materials and Methods. After model validation, PK11195 (yellow) and ND-PK (light blue) were docked into the CAR ligand-binding pocket (A). Interactions of PK11195 (B), ND-PK (C), CINPA1 (D), and CITCO (E) with residues in the ligand-binding pocket of hCAR. Residues are color-coded as follows: dark blue, interact with all structures; light blue, PK11195 and ND-PK; orange, PK11195, CINPA1, and CITCO; red, PK11195 and CINPA1; green, CINPA1 and CITCO; yellow, CITCO only; and purple, ND-PK only.
Discussion
The biologic function of CAR is regulated by the interplay between specific cellular factors and small molecular modulators. PK11195, a well-known peripheral benzodiazepine receptor ligand, has been used as a potent hCAR deactivator in cell-based luciferase reporter assays. In contrast to CAR antagonism exhibited in immortalized cell lines, PK11195 robustly induces the expression of both CYP2B6 and CYP3A4 in HPHs, which are prototypical targets for hCAR and hPXR, respectively. The mechanistic basis for this observed discrepancy is largely unknown, although it was presumed that PK11195-mediated P450 induction in HPHs was attributed to its activation of hPXR. Here, we show that PK11195 significantly induces the expression of CYP2B6 and CYP3A4 in PXR-KO HepaRG cells, which demonstrates that PK11195 can stimulate P450 expression independent of PXR. Utilizing a HPH-HepG2 coculture model, we show that introduction of metabolically competent HPHs is sufficient to convert PK11195 from an antagonist to an agonist of hCAR in HepG2 cells. In HPHs, PK11195 is biotransformed to ND-PK and COOH-PK. Further studies demonstrated that ND-PK is the active metabolite that potently activates hCAR and induces the expression of CYP2B6 and CYP3A4. Moreover, structure-activity analysis reveals that N-demethylation of PK11195 allows its side chain to rotate away from residues in the CAR ligand-binding pocket toward a conformation in favor of CAR activation.
CAR and PXR regulate an overlapping array of target genes and share many common chemical modulators (Hernandez et al., 2009; Mackowiak and Wang, 2016). Although such crosstalk between CAR and PXR can be beneficial by forming a defensive network against xenobiotics, it makes the delineation of specific function of each individual receptor extremely challenging, particularly in cells such as HPHs, where both CAR and PXR are abundant and functionally intact. Recently, the HepaRG cell line has emerged as a useful alternative for HPHs; differentiated HepaRG cells exhibit prototypical HPH morphology, inductive expression of major drug-metabolizing enzymes and transporters, and have been used for in vitro drug metabolism and toxicology studies (Jossé et al., 2008; Andersson et al., 2012). The PXR-KO HepaRG cell line provides an excellent model to determine the contribution of CAR/PXR to PK11195-mediated P450 induction. Our results uncover an unexpected induction of both CYP2B6 and CYP3A4 by PK11195 in the PXR-KO HepaRG cells, although induction by selective hPXR activator RIF was fully abrogated. These findings provide conclusive evidence that PK11195 can induce CYP2B6/CYP3A4 expression in physiologically relevant hepatic cells independent of hPXR. Indeed, previous and current studies in HPHs have shown that PK11195 induces both CYP2B6 and CYP3A4 in a pattern that mimics that of PB, a dual activator of hCAR and hPXR (Li et al., 2008; Anderson et al., 2011). Together, these results indicate that PK11195 modulates CAR differently in HPHs and HepaRG cells versus in immortalized cell lines, and is most likely metabolized from an antagonist to an agonist in cells exhibiting physiologically relevant metabolism.
Lack of metabolism capacity is a significant drawback associated with the use of immortalized cell lines in toxicity assessment and drug development. Cell-based luciferase reporter assays using immortalized cell lines in particular have been extensively used to investigate nuclear receptor activity and predict target gene expression. However, proper interpretation of such data has become a heightened concern in both academia and the pharmaceutical industry. Several lines of evidence indicate that introduction of metabolic capacity to cell cultures appears to be an attractive solution to overcome this issue. In this regard, we have previously used an HPH-leukemia/lymphoma cell coculture model to show that the presence of HPHs markedly increases the biotransformation of cyclophosphamide, a chemotherapeutic prodrug, to its pharmacologically active metabolite and leads to enhanced anticancer activity in cocultured HL-60 and SU-DHL-4 cells (Wang et al., 2013; Hedrich et al., 2016). Using a HPH-HepG2 coculture system in the current study, we observed that PK11195 concentration dependently increased CAR activation in contrast to decreasing CAR activity when exposed to HepG2 cells only. More importantly, such agonistic effects of PK11195 in the coculture can be reversed by KET, suggesting that CYP3A4 plays a key role in the metabolism-based conversion of PK11195 in HPHs. It is not uncommon that metabolism can influence the pharmacological action of drugs. For instance, chrysin, a dietary flavonoid, markedly induces the expression and activity of UDP-glucuronosyltransferase 1A1 in HepG2 and Caco-2 cells, but not in HPHs (Smith et al., 2005). Buprenorphine, a potent activator of PXR in HepG2 cells, is not a physiologically relevant activator of PXR or an inducer of associated P450s in HPHs (Li et al., 2010). On the other hand, phenytoin, an antiepileptic agent, is a potent inducer of CYP2B6 and CYP3A4 in HPHs but does not activate CAR or PXR in cell-based reporter assays (Wang et al., 2004). Collectively, these studies demonstrate that the metabolic capacity of a cellular system can be a key determinant for the biologic function of a given compound, including its role in nuclear receptor activation.
Previous reports have postulated that PK11195 is rapidly biotransformed into two major metabolites: the N-desmethyl metabolite, ND-PK, and the amide hydrolysis product, COOH-PK (Roivainen et al., 2009). In the current study, we have evaluated the activation of hCAR and induction of related P450s by both metabolites. Notably, ND-PK significantly increased the luciferase activity of the CYP2B6 reporter by activating hCAR or hCAR1+A in HepG2 cells and induced the expression of CYP2B6 and CYP3A4 in HPHs, HepaRG, and PXR-KO HepaRG cells. In contrast, COOH-PK failed to activate hCAR in HepG2 cells and only marginally induced CYP2B6/CYP3A4 expression in HPHs. Consistent with these observations fluorescent microscopy analysis of adenovirus-expressing EYFP-hCAR-infected HPHs further confirmed that ND-PK but not COOH-PK efficiently translocates CAR from the cytoplasm to the nucleus of HPH, the first step in CAR activation. This discovery may also provide a mechanism for the previously observed PK11195-mediated CAR nuclear translocation in HPHs (Li et al., 2009). Together, these findings suggest that removing one methyl group from PK11195 changes it from an antagonist to an agonist of CAR and contributes to P450 induction in HPHs.
Mechanistically, ligand binding changes the secondary structure of a nuclear receptor, influences the recruitment of coregulators, and alters the target gene expression thereafter. Previous reports have indicated that nuclear localized CAR can interact with coactivators such as SRC-1 and GRIP1 without the presence of agonists (Muangmoonchai et al., 2001; Min et al., 2002). Our mammalian two-hybrid results demonstrate that PK11195-repressed CAR/SRC-1 interaction can be efficiently rescued by ND-PK. However, this recovery is moderate in the CAR/GRIP1 interaction, reflecting the differential capacity between PK11195 and ND-PK in influencing the recruitment of SRC-1 versus GRIP1 by CAR.
Computational modeling studies have shown that the constitutive activity of CAR is mediated by residues in the binding pocket interacting with and stabilizing the activation function 2 (AF2) domain (Andersin et al., 2003; Xu et al., 2004; Windshügel et al., 2005). Agonists tend to bind and further stabilize the AF2 domain in the active conformation, while antagonists disrupt its stability (Jyrkkärinne et al., 2008). To explore how the N-demethylation of PK11195 has such a drastic effect on CAR activity, we used docking studies to probe the structure-activity relationship of PK11195 and ND-PK with CAR. Although both compounds bound in similar conformations, PK11195 interacted with residues important to CAR activation, such as V199, H203, Y224, and Y326, while ND-PK does not. Residues V199 and Y326 are thought to stabilize the CAR AF2 domain in the active conformation by interacting with H12, while Y224 may be involved in local protein folding. Mutating any of these residues abrogates the basal activity of CAR while mutating H203 reduces CAR activity by 50%, demonstrating the importance of these residues to CAR activity (Jyrkkärinne et al., 2005). Therefore, PK11195 interactions with these residues may destabilize the AF2 domain, while loss of the N-methyl group in ND-PK could restabilize H12. Indeed, the potent CAR antagonist CINPA1 also interacts with these residues, suggesting such interactions are important in CAR antagonism (Cherian et al., 2016). CITCO interacts with V199, Y224, and Y326, but the interaction distances are generally greater than those of PK11195 or CINPA1 and thus may not displace these residues enough to inhibit stabilization of H12 (Supplemental Table 1). Although docking studies provide a possible mechanism for this drastic change in CAR activity, future studies will use molecular dynamics and mutagenesis to further probe the detailed structure-activity relationship between PK11195 and ND-PK with CAR.
In conclusion, this study demonstrates that PK11195 is metabolically converted from an antagonist to an agonist of hCAR and this conversion contributes significantly to the observed induction of CYP2B6 and CYP3A4 in HPHs and HepaRG cells. We show that ND-PK is the metabolite responsible for PK11195-mediated CAR activation by facilitating CAR interactions with SRC-1 and GRIP1 and enhancing CAR nuclear translocation in HPHs. The demethylation of PK11195 also disrupts its interaction with residues critical to CAR activity, providing a possible mechanism for the activity shift. Additionally, this report highlights the importance of metabolic competence when attempting to identify modulators of nuclear receptors and provides a possible solution to this problem with a novel HPH coculture system.
Acknowledgments
The authors thank Michael Mitchell from Sigma Life Sciences for kindly providing the PXR-KO-HepaRG cell line. The LC-MS analysis of PK11195 metabolism was performed in the Mass Spectrometry Center at the University of Maryland School of Pharmacy.
Authorship Contributions
Participated in research design: Mackowiak, L. Li, Welch, D. Li, Jones, Kane, Swaan, Wang.
Conducted experiments: Mackowiak, L. Li, Welch, Jones, Heyward.
Contributed new reagents or analytic tools: Heyward.
Performed data analysis: Mackowiak, L. Li, Welch, Jones, Kane, Swaan, Wang.
Wrote or contributed to the writing of the manuscript: Mackowiak, L. Li, Welch, Jones, Kane, Swaan, Wang.
Footnotes
- Received February 20, 2017.
- Accepted April 20, 2017.
This work was supported by the National Institutes of Health National Institute of General Medicine [Grant R01 GM107058]. B.M. and M.A.W. are partly supported by The University of Maryland’s Center of Excellence in Regulatory Science and Innovation (M-CERSI) Scholars Program funded by the Food and Drug Administration [Grant 1U01FD005946].
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- AF2
- activation function 2
- CAR
- constitutive androstane receptor
- CINPA1
- constitutive androstane receptor inhibitor not pregnane X receptor activator 1
- CITCO
- 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime
- COOH-PK
- 1-(2-chlorophenyl)isoquinoline-3-carboxylic acid
- DMSO
- dimethylsulfoxide
- EYFP-hCAR
- enhanced yellow fluorescent protein-tagged human constitutive androstane receptor
- hCAR
- human constitutive androstane receptor
- HPH
- human primary hepatocyte
- hPXR
- human pregnane X receptor
- KET
- ketoconazole
- KO
- knockout
- LC
- liquid chromatography
- MS
- mass spectrometry
- ND-PK
- (R)-N-desmethyl 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide
- P450
- cytochrome P450
- PB
- phenobarbital
- PCR
- polymerase chain reaction
- PK11195
- 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide
- PXR
- pregnane X receptor
- RIF
- rifampicin
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}