


Abstract
As a promiscuous xenobiotic sensor, the constitutive androstane receptor (CAR; NR1I3) regulates the expression of multiple drug-metabolizing enzymes and transporters in liver. The constitutively activated nature of CAR in the cell-based transfection assays has hindered its use as a predictor of metabolism-based drug-drug interactions. Here, we have identified 1-(2-chlorophenylmethylpropyl)-3-isoquinoline-carboxamide (PK11195), a typical peripheral benzodiazepine receptor (PBR) ligand, as a selective and potent inhibitor of human (h) CAR. In cell-based transfection assays, PK11195 inhibited the constitutive activity of hCAR more than 80% at the concentration of 10 μM, and the PK11195-inhibited activity was efficiently reactivated by the direct CAR activator, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl) oxime, but not by the indirect hCAR activator, phenobarbital. Mammalian two-hybrid and GST pull-down assays showed that PK11195 repressed the interactions of hCAR with the coactivators steroid receptor coactivator-1 and glucocorticoid receptor-interacting protein 1 to inhibit hCAR activity. The inhibition by PK11195 specifically occurred to the hCAR: PK1195 strongly activated human pregnane X receptor (PXR), whereas it did not alter the activity of the mouse CAR and mouse PXR. In addition, PBR played no role in the PK11195 inhibition of hCAR because the inhibition fully occurred in the HeLa cells in which the PBR was knocked down by small interfering RNA. In the Car-/- mouse liver, PK11195 translocated enhanced yellow fluorescent protein-hCAR into the nucleus. These results are consistent with the conclusion that PK11195 is a novel hCAR-specific antagonist that represses the CAR-coactivator interactions to inhibit the receptor activity inside the nucleus. Thus, PK11195 can be used as a chemical tool for studying the molecular basis of CAR function.
The constitutive androstane receptor (CAR, NR1I3) is an important transcription factor that transfers endogenous and exogenous stimuli into cellular responses by regulating the expression of its target genes that code for cytochrome P450 (P450) and transport proteins (Sueyoshi et al., 1999; Tzameli et al., 2000; Ueda et al., 2002; Huang et al., 2004; Maglich et al., 2004). Thus, CAR organizes a cellular defense system against xenobiotic insults by increasing hepatic capacity for metabolism and excretion. Besides its role in drug metabolism, CAR is also reported to have the ability to regulate various hepatic functions, including gluconeogenesis, fatty acid oxidation, and the metabolism of steroid hormones and bilirubin (Sugatani et al., 2001; Huang et al., 2003; Kodama et al., 2004; Ueda et al., 2005). Moreover, CAR promotes the development of hepatocellular carcinoma in phenobarbital (PB)-treated mice (Yamamoto et al., 2004; Huang et al., 2005).

PK11195 deactivates constitutive activity of hCAR in HepG2 cells. A, structure of PK11195. B, HepG2 cells were transfected with CYP2B6-2.2 kb reporter and hCAR expression vectors. Transfected cells were treated with PK11195, CITCO, or PB at indicated concentrations for 24 h. Luciferase activities were determined and expressed relative to vehicle control (CT). Data represent the mean ± S.D. (n = 3) (**, P < 0.01 denotes comparison with DMSO group; #, P < 0.05, and ##, P < 0.01 denote comparison with 10 μM PK11195 group).
Although the importance of CAR in the regulation of liver function becomes clear, the molecular mechanism underlying CAR activation is still elusive. In contrast to most NRs, CAR displays high constitutive activity in the absence of agonistic ligand binding (Baes et al., 1994). CAR is also characterized as an NR that could be activated by either direct ligand binding, such as 6-(4-chlorophenyl)imidazo [2,1-b][1,3] thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl) oxime (CITCO) (Maglich et al., 2004), or indirect activation, such as PB (Honkakoski et al., 2003; Maglich et al., 2004). In intact liver, CAR is localized predominantly in the cytoplasm in the absence of CAR activators but is accumulated in the nucleus after exposure to PB-type compounds (Kawamoto et al., 1999; Wang et al., 2004). Thus, nuclear translocation of CAR has been established as the initial step for CAR-mediated transcriptional activation. In contrast, this nuclear receptor exhibits a high level of constitutive transcriptional activity in immortalized cells and accumulates spontaneously in nuclei. This characteristic of CAR in cell lines has become one of the major difficulties in elucidating the mechanism underlying CAR activation. Alternatively, identification of highly effective CAR antagonists would provide novel tools to study the molecular basis of CAR function and offer new molecules with potential clinical applications. Although hCAR exhibits several common characteristics with its rodent counterparts, there are distinct biochemical differences between rodent and hCAR. For example, TCPOBOP can bind and activate mouse but not human CAR, and the androstane metabolites androstanol and androstenol repress the constitutive activity of mouse CAR in HepG2 cells, but have only marginal effect on hCAR (Forman et al., 1998; Tzameli et al., 2000). Moreover, rodent but not hCAR is susceptible to calcium/calmodulin-dependent kinase-mediated inhibition of PB induction of CYP2B expression and CAR transactivation of phenobarbital-responsive enhancer module (PBREM) (Kawamoto et al., 2000; Yamamoto et al., 2003). Among the few known antagonistic modulators of hCAR, the effect of clotrimazole (CLZ) on the constitutive activity of hCAR is contradictory [ranging from repressing (Tzameli et al., 2000) to no effect (Toell et al., 2002) and to activating (Jyrkkärinne et al., 2005)] in different cell lines. Meclizine has been reported to act as a moderate inverse agonist of hCAR, which inhibits the hCAR activity approximately 50% in cell-based transfection assays (Huang et al., 2004).
Here, we have found that PK11195, the well-known PBR ligand, is a specific and potent antagonist of hCAR in both HepG2 cells and human primary hepatocytes. Moreover, we used small interfering RNA (siRNA) knock-down, mammalian two-hybrid, GST pull-down, and in situ expression of hCAR in the Car-/- mouse liver to further investigate the molecular mechanism by which PK11195 inhibits the hCAR activity. Overall, the identification of PK11195 as a novel functional antagonist of human CAR provides a useful chemical tool for studying the molecular basis of hCAR function.
Materials and Methods
Chemicals and Biological Reagents. PK11195, PB, rifampicin (RIF), Picrotoxin, Ro5-4864, FGIN-1-27, carbamezapine (CMZ), and sulforaphane (SFN) were purchased from Sigma-Aldrich (St. Louis, MO). CITCO was obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). Oligonucleotide primers and TaqMan fluorescent probes were synthesized by Sigma Genosys (The Woodlands, TX) and Applied Biosystems (Foster City, CA), respectively. The Dual-Luciferase Reporter Assay System was purchased through Promega (Madison, WI). siGENOME Duplex specific for PBR or Non-Target was synthesized by Dharmacon (siRNA-BPR and siRNA-NT). Matrigel; insulin; and insulin/transferrin/selenium were obtained from BD Biosciences (Bedford, MA). Other cell culture reagents were purchased from Invitrogen (Carlsbad, CA) or Sigma-Aldrich.

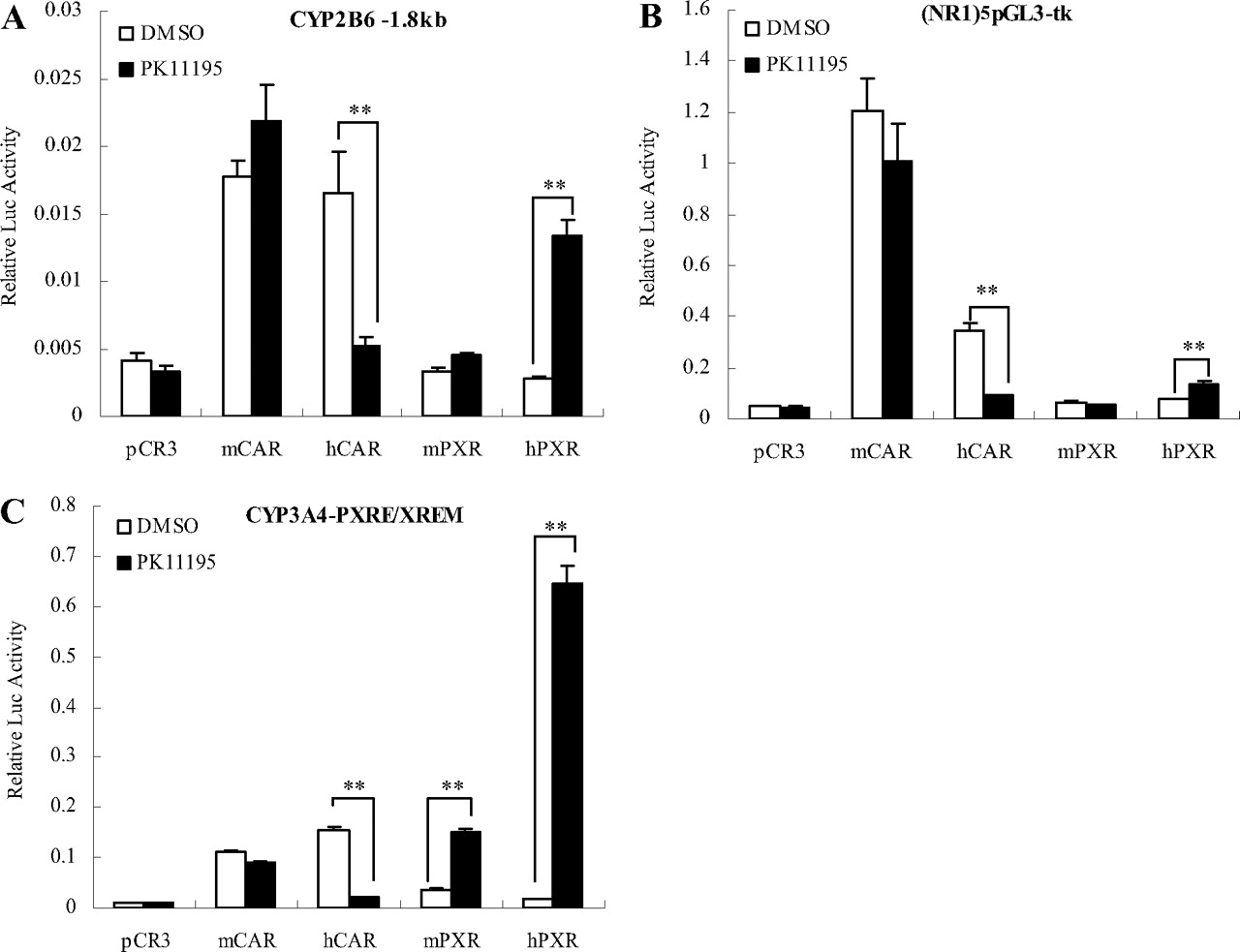
PK11195 selectively inhibited the constitutive activity of hCAR. HepG2 cells were transfected with CYP2B6-1.8 kb (A), (NR1)5pGL3-tk (B), or CYP3A4-PXRE/XREM (C) reporter construct in the presence of mCAR, hCAR, mPXR, or hPXR expression vectors. Transfected cells were then treated with PK11195 (10 μM) or 0.1% DMSO as CT. Dual luciferase activities were determined and expressed relative to CT. All data are presented as mean ± S.D. (n = 3) (**, p < 0.01).
Plasmid Constructions. The pCR3-hCAR, hCAR/pEYFP-c1, and (NR1)5pGL3-tk were generated as described previously (Sueyoshi et al., 1999, Kawamoto et al., 1999; Zelko et al., 2001). Plasmids used for mammalian two-hybrid assay, pG5-Luc, pACT, and pBind were obtained from Promega. pACT-hPXR, pM-SRC-1 (621-765), and pBind-SMRT (981-1450) were described in previous reports (Chang et al., 1999; Ueda et al., 2005). Human CAR full-length coding DNA was cloned into BamHI and XbaI sites of pACT (pACT-hCAR). Plasmids for GST pull-down assay, pcDNA3.1-hGRIP1, and pcDNA3.1-mSRC-1 were described previously (Ueda et al., 2002; Hosseinpour et al., 2006). The pSG5-hPXR expression vector and the CYP3A4-PXR response element (PXRE)/XREM luciferase reporter construct [p3A4-362(7836/7208ins)] were obtained from Drs. Steven Kliewer (University of Texas Southwestern Medical Center, Dallas, TX) and Bryan Goodwin (GlaxoSmithKline, Research Triangle Park, NC), respectively. The CYP2B6 reporter constructs, containing the proximal PBREM (CYP2B6-1.8 kb) or both PBREM and the distal XREM (CYP2B6-2.2 kb) were generated as described previously (Sueyoshi et al., 1999; Wang et al., 2003). The pRL-TK Renilla reniformis luciferase plasmid used to normalize firefly luciferase activities was from Promega.
Transfection Assays in Cell Lines. HepG2 or HeLa cells in 24-well plates were transfected with hCAR or hPXR expression vector, and CYP2B6-2.2 kb, CYP2B6-1.8 kb, (NR1)5pGL3-tk, or CYP3A4-PXRE/XREM reporter constructs using Fugene 6 Transfection Kit according to the manufacturer's instruction. Twenty-four hours after transfection, cells were treated with solvent (0.1% DMSO) or test compounds at the concentration of PK11195 (10 μM), picrotoxin (10 μM), Ro5-4864 (10 μM), FGIN-1-27 (10 μM), CMZ (30 μM), CITCO (1 μM), or RIF (10 μM) for 24 h. Cell lysates were assayed for firefly activities normalized against the activities of cotransfected R. reniformis luciferase using the Dual-Luciferase Kit (Promega, WI). Data are represented as mean ± S.D. of three individual transfections. To analyze PXR- and CAR-mediated CYP3A4 expression in HepG2 and Huh7 cells, cells seeded in 12-well plates were transfected with hCAR or hPXR expression vectors and then treated with PK11195 (10 μM), CITCO (1 μM), or RIF (10 μM) for 24 h. Total RNA was isolated using RNeasy Mini Kit (QIAGEN, Valencia, CA).
Human Primary Hepatocytes Cultures and Treatments. Liver tissues were obtained by qualified medical staff after donor consent and prior approval from the Institutional Review Board at the University of Maryland at Baltimore. Hepatocytes were isolated from human liver specimens by a modification of the two-step collagenase digestion method as described previously (LeCluyse et al., 2005). Hepatocytes were seeded at 1.5 × 106 cells/well in six-well BioCoat plates in Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 4 μg/ml insulin, and 1 μM dexamethasone. After 4 to 6 h of attachment at 37°C in a humidified atmosphere of 5% CO2, cells were overlaid with Matrigel (0.25 mg/ml) in Williams' E medium supplemented with insulin, transferrin, and selenium, 0.1 μM dexamethasone, 100 U/ml penicillin, and 100 μg/ml streptomycin. The hepatocytes were maintained for 36 h before the treatment with PK11195 (10 μM), SFN (25 μM), RIF (10 μM), CITCO (1 μM), or cotreatment of SFN with PK11195, RIF, or CITCO, respectively, for another 24 h.

Induction of CYP2B6 and CYP3A4 expression in human primary hepatocyte cultures. Human hepatocytes (UM-HL-001 and UM-HL-002) cultured in Williams' E medium were treated for 24 h with 10 μM PK11195, 25 μM SFN, 10 μM picrotoxin, 10 μM Ro5-4868, 10 μM FGIN-1-29, and 30 μM CMZ, as well as cotreatment of SFN with PK11195, RIF, and CITCO, respectively. Total RNA was collected, reverse-transcribed, and subjected to TaqMan real-time PCR. CYP2B6 and CYP3A4 expression levels were normalized against β-actin. Induction of CYP2B6 and CYP3A4 relative to control was calculated as described under Materials and Methods. All data are presented as mean ± S.D. (n = 3) (*, p < 0.05; **, p < 0.01).
Determination of CYP3A4 and CYP2B6 mRNA Expressions by Real-Time PCR Analysis. Total RNA was isolated using the RNeasy Mini Kit (QIAGEN) and reverse-transcribed using High-Capacity cDNA Archive kit (Applied Biosystems) according to the manufacturers' instructions. Primers and probes for CYP2B6 and CYP3A4 mRNA detection are as follows, in the order of forward primer, probe, and reverse primer: CYP2B6 1299 to 1366 base pairs, 5-AAGCGGATTTGTCTTGGTGAA-3, 6-FAM-CATCGCCCGTGCGGAATTGTTC-TAMRA, and 5-TGGAGGATGGTGGTGAAGAAG-3; and CYP3A4 59 to 179 base pairs, 5-TCAGCCTGGTGCTCCTCTATCTAT-3, 6-FAM-TCCAGGGCCCACACCTCTGCCT-TAMRA, and 5-AAGCCCTTATGGTAGGACAAAATATTT-3. CYP2B6 or CYP3A4 mRNA expression was normalized against that of human β-actin, which was detected using a predeveloped primer/probe mixture (Applied Biosystems). Multiplexed TaqMan PCR assays were performed in 96-well optical plates on an ABI Prism 7000 Sequence Detection System (Applied Biosystems). -Fold induction values were calculated according to the equation 2ΔΔCt, where ΔCt represents the differences in cycle threshold numbers between the target gene and β-actin, and ΔΔCt represents the relative change in these differences between control and treatment groups.
siRNA Knockdown of the PBR in HeLa Cells. The siRNA specific for PBR and a Non-Targeting siRNA were obtained from Dharmacon. To detect knockdown of endogenous PBR, HeLa cells plated in 12-well plates were transfected with siRNA-PBR (40 pmol) or siRNA-NT (40 pmol) using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, cells were harvested, and total RNA was isolated and reverse-transcribed into cDNA as described above. PBR gene expression was measured using SYBR real-time PCR with specific primers for PBR (forward, 5-CTTCTTTGGTGCCCGACAAAT-3, and reverse, 5-GCCATACGCAGTAGTTGAGTG-3) and normalized against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase. Detection of the PCR products was done with the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Gene knockdown were determined by calculating the 2ΔΔCt values as described above. PCR products (15 μl each), at 29 cycles for PBR and 26 cycles for glyceraldehyde-3-phosphate dehydrogenase, were also subjected to electrophoresis in 2% agarose gel. In cell-based reporter assays, after 24 h of siRNA transfection, hCAR expression vector and CYP2B6-2.2 kb reporter vector were also transfected in HeLa cells. The double-transfected cells were treated with PK11195 (10 μM) or CITCO (1 μM) for 24 h and then subjected to dual-luciferase assays as described above.
In Vivo Gene Transfection and Confocal Microscopy.Car-/- mice, as described previously (Ueda et al., 2002), were housed in a pathogen-free animal facility with standard 12-h light/dark cycles and provided autoclaved rodent chow and drinking water ad libitum. Animals weighing 18 to 20 g were used for hCAR translocation studies. In hCAR localization experiments, Car-/- mice were injected with 10 μg of EYFP-hCAR expression plasmid using the tail vein injection method (Sueyoshi et al., 2002; Wang et al., 2004). Treatment occurred at 3 h after gene delivery by intraperitoneal injection of vehicle (DMSO, CT), 15 mg/kg PK11195, or 100 mg/kg PB. Mouse livers were collected 8 h after the plasmid injection, embedded into Tissue-Tek OTC, and immediately frozen. Microscopic analysis of frozen liver sections was performed as described previously (Zelko et al., 2001). EYFP-hCAR was visualized in hepatocytes using a Zeiss LSM510 confocal laser scanning microscope (Carl Zeiss GmbH, Jena, Germany) at an excitation wavelength of 514 nm. For each treatment group, approximately 100 mouse hepatocytes expressing EYFP-hCAR were counted per mouse liver and classified according to cytosolic, nuclear, or mixed (cytosolic and nuclear) hCAR localization. Livers from two mice were evaluated by confocal analysis for each treatment.
Mammalian Two-Hybrid Assays. COS1 cells seeded in 24-well plates were transfected with 110 ng of the reporter gene plasmid pG5luc, 40 ng of expression plasmids encoding GAL4-DBD/coregulator fusions, 80 ng of expression plasmids encoding the respective VP16-AD/hCAR fusions, and 20 ng of reference plasmid pRL-TK each well using Fugene 6 (Roche, Indianapolis, IN). Twenty-four hours after transfection, the cells were treated with CITCO (1 μM), PK11195 (10 μM), RIF (10 μM), or vehicle control (0.1% DMSO) for 24 h. Luciferase activities were measured in cell lysates using the Dual-Luciferase kit (Promega) as described above. Data represent the mean ± S.D. of three individual transfections.
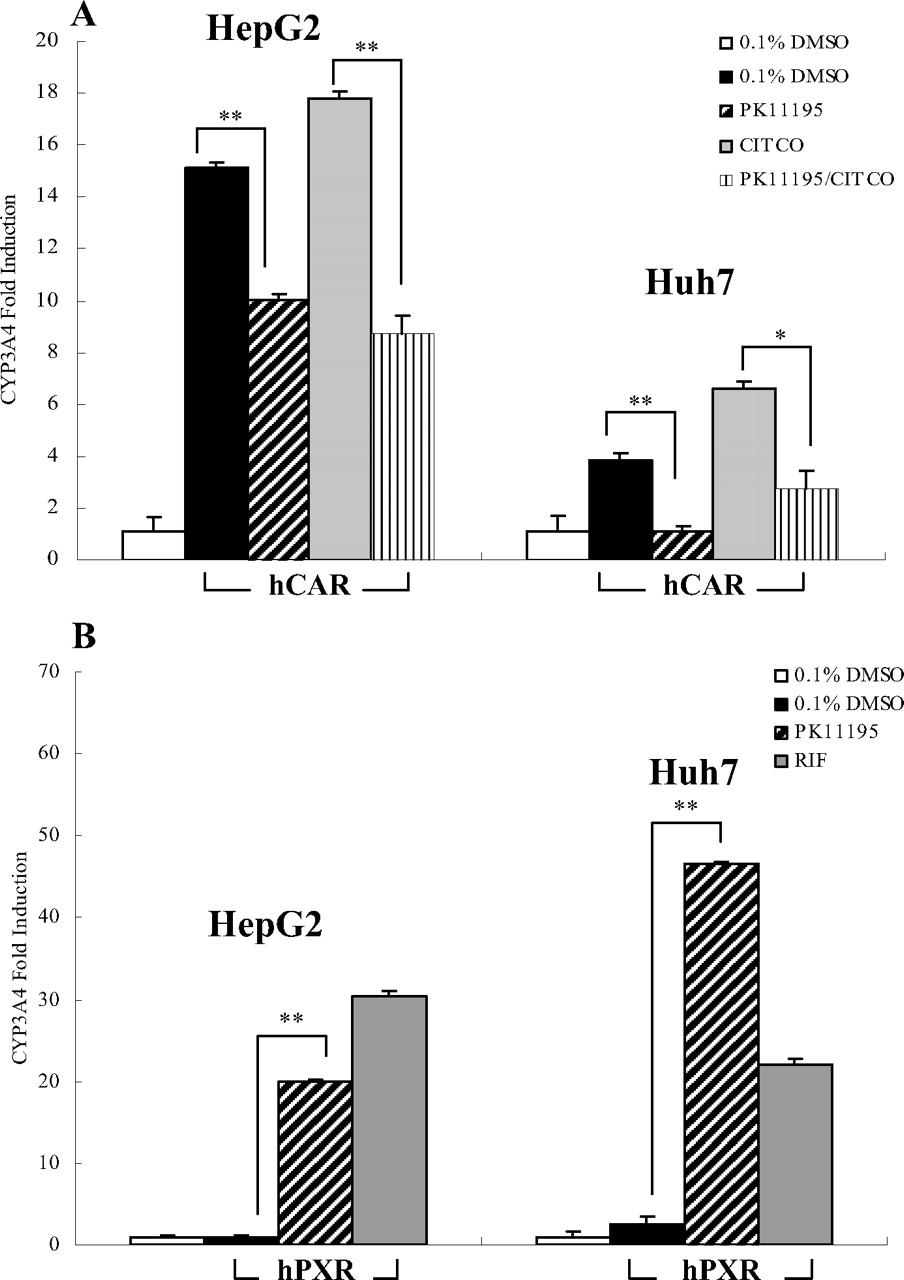
The effect of PK11195 on hCAR- or hPXR-induced expression of CYP3A4. HepG2 and Huh7 cells were transfected with hCAR (A) or hPXR (B) expression vectors, and treated with PK11195 (10 μM), CITCO (1 μM), or RIF (10 μM) for 24 h. Total RNA was collected, reverse-transcribed, and subjected to TaqMan real-time PCR as described under Materials and Methods. All data are expressed as mean ± S.D. (n = 3), (*, p < 0.05; **, p < 0.01).
GST Pull-Down Assay. The GST-hCAR fusion protein was expressed in Escherichia coli strain BL21 cells and purified using glutathione-Sepharose 4B (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). 35S-labeled SRC-1 and GRIP-1 were produced from pcDNA3.1-mSRC1 and pcDNA3.1-hGRIP1 using the TNT T7 quick-coupled transcription/translation system (Promega) along with 35S-labeled methionine. GST-hCAR or GST coupled to the glutathione-Sepharose 4B was incubated with the 35S-labeled SRC-1 or GRIP-1 in 50 mM HEPES buffer, pH 7.5, containing 0.1 M NaCl and 0.1% Triton X-100 for 20 min at room temperature in the presence of DMSO (0.1%), PK11195 (100 μM), CITCO (1 μM), or CITCO (1 μM) + PK11195 (100 μM). The resin was then recovered by centrifugation and washed three times in the same buffer. Proteins were extracted from the resin by heating for 10 min at 70°C in NuPAGE LDS sample buffer (Invitrogen) and separated on 4-10% BisTris gel in NuPAGE MOPS SDS running buffer for 1 h at 150 V. The gel was then dried under vacuum, and proteins were detected by autoradiography.
Statistical Analysis. All data represent at least three independent experiments and are expressed as the mean ± S.D. Statistical comparisons were made using Students' t test. Statistical significance was set at p values <0.05 (*) or <0.01 (**).
Results
Inhibition of the hCAR's Constitutive Activity by PK11195. First, we detected the effect of PK11195 on the constitutive activity of hCAR by transfection assay in HepG2 cells. It was found that PK11195 could efficiently reduce hCAR activity in a dose-dependent manner. Compared with vehicle control, the luciferase activities of CYP2B6-2.2 kb reporter construct decreased by 85% after PK11195 (10 μM) treatment (Fig. 1B). In a similar experiment, transfected HepG2 cells were cotreated with PK11195 and a variety of hCAR activators. It is interesting that only the direct activator of hCAR, CITCO, reversed the inhibitory effect of PK11195, whereas PB, the typical indirect activator of hCAR, had no effect on the PK11195-mediated deactivation (Fig. 1B). This result suggests that PK11195 might bind specifically to the ligand binding domain of hCAR.
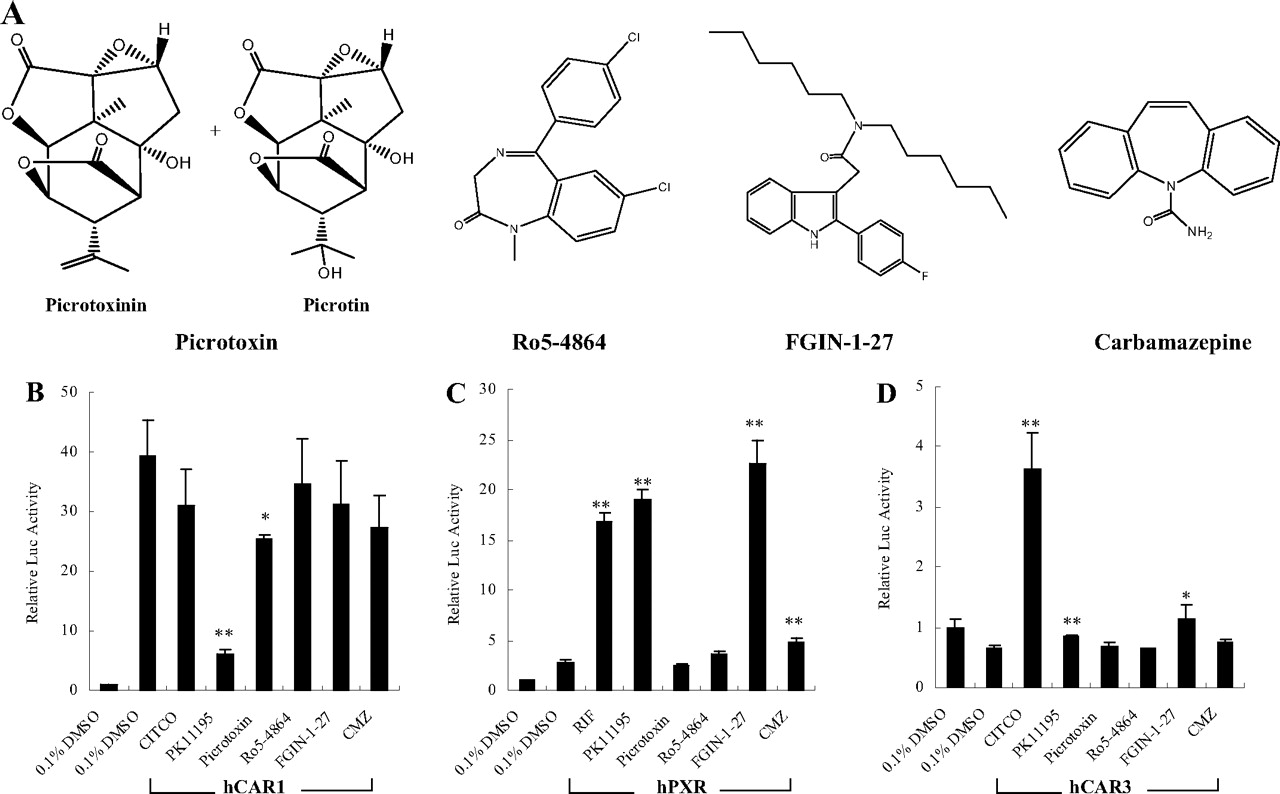
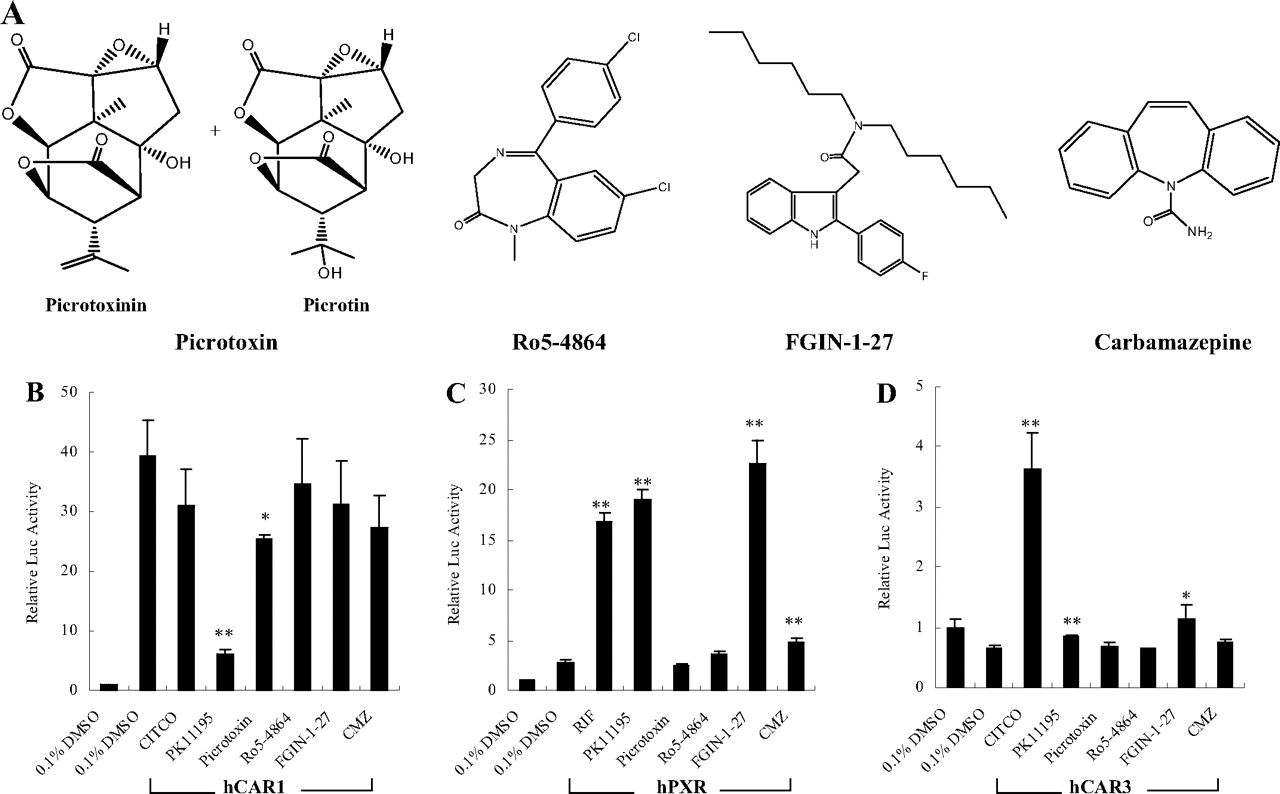
The effects of PBR ligands on the activity of hCAR and hPXR. Four structurally different PBR ligands (A) were selected in this reporter assay. HepG2 cells were transfected with hCAR (B), hPXR (C), or hCAR3 (D) expression vectors in the presence of CYP2B6-2.2 kb reporter construct. Transfected cells were then treated with PBR ligands (10 μM PK11195, 10 μM picrotoxin, 10 μM Ro5-4868, 10 μM FGIN-1-27, or 30 μM CMZ) for 24 h. CITCO (1 μM) and RIF (10 μM) were used as positive control for hCAR and hPXR activation, respectively. Luciferase activities were determined and expressed relative to CT. Data represent the mean ± S.D. (n = 3) (*, p < 0.05; **, p < 0.01).
The Effect of PK11195 on the Activities of Other Nuclear Receptors. Because a significant species-specific difference exists between hCAR and its rodent counterparts, expression vectors of mCAR, hCAR, mPXR, and hPXR were used to determine the inhibitory specificity of PK11195. HepG2 cells were transfected with CYP2B6 luciferase reporter constructs containing PBREM (CYP2B6-1.8 kb), five-repeat of the NR1 (NR1)5pGL3-tk, or CYP3A4-PXRE/XREM in the presence of different CAR and PXR expression vectors. As demonstrated in Fig. 2, hCAR activity was significantly suppressed by PK11195 in assays with all three reporter constructs. In contrast, only negligible activity changes of mouse CAR was observed after treatment with PK11195. It is noteworthy that PK11195 exhibits potent activation of human PXR through all three reporter constructs, whereas moderate activation of mPXR was observed only with the CYP3A4 construct (Fig. 2C).
PK11195 Induction of CYP2B6 and CYP3A4 Gene Expression in Human Primary Hepatocytes. Given the fact that human CAR and PXR share several common target genes such as CYP2B6 and CYP3A4, we further evaluated the effects of PK11195 on the expression of CYP2B6 and CYP3A4 genes in human primary hepatocyte cultures. Human hepatocytes from two different livers were treated with PK11195, CITCO, or RIF in the presence or absence of a newly identified hPXR deactivator SFN as described under Materials and Methods. In agreement with previous reports, RIF efficiently induced both CYP2B6 and CYP3A4, whereas CITCO exhibited marked preferential induction of CYP2B6 over CYP3A4 in both hepatocyte cultures (Fig. 3, A and B) (Faucette et al., 2006, 2007). It is noteworthy that 10 μM PK11195 strongly induced the mRNA expression of both CYP2B6 and CYP3A4 in treated hepatocytes, and these inductions were significantly repressed by the cotreatment of 25 μM SFN. Concomitant treatment with SFN (25 μM) and RIF nearly abolished the induction of CYP3A4 and CYP2B6 by RIF alone (Fig. 3, A and B), consistent with a previous report (Zhou et al., 2007). In contrast, induction of CYP2B6 by the selective hCAR activator CITCO was only moderately repressed by SFN treatment. Together, these observations suggest that PK11195 induces P450 expression in human primary hepatocytes via the activation of endogenous PXR.
PK11195 Inhibits the hCAR-Mediated Endogenous CYP3A4 Expression. Because growing evidence demonstrates that both CAR and PXR regulate CYP3A4 gene expression (Xie et al., 2000), we further tested the distinct roles of PK11195 on hCAR and hPXR in HepG2 and Huh7 cells. Cells were transfected with hCAR or hPXR expression vectors, and as expected, transfection of hCAR in HepG2 and Huh7 cells enhanced the basal expression of CYP3A4 gene as a result of the constitutive activation of hCAR in immortalized cell lines. Treatment of PK11195 resulted in significant repression of both basal and CITCO-induced CYP3A4 mRNA expression levels (Fig. 4A). Consistent with the results from our cell-based reporter assays (Fig. 2), PK11195 also induced CYP3A4 mRNA expressions in hPXR-transfected HepG2 and Huh7 cells (Fig. 4B).

PK11195 deactivation of hCAR is independent of PBR. HeLa cells were transfected with siRNA-NT or siRNA-PBR. Forty-eight hours after transfection, down-regulation of PBR gene was detected by real-time PCR (A). In a parallel experiment, HeLa cells were first subjected to the specific siRNA-PBR for 24 h, and the cells were subsequently transfected with hCAR expression vector and CYP2B6-2.2 kb reporter vector (B). The transfected cells were then treated with PK11195 (10 μM) or CITCO (1 μM). Luciferase activities were determined and expressed relative to CT. Data represent the mean ± S.D. (n = 3) (**, p < 0.01).
The Effects of PBR Ligands on the Activation of hCAR and hPXR. Other than PK11195, a number of PBR ligands with diverse chemical structures have been identified previously (Roberge et al., 2004). To examine whether other PBR ligands exhibit similar effects on the activation/deactivation of hCAR and hPXR, four known PBR ligands, picrotoxin, Ro5-4864, FGIN-1-27, and CMZ (Fig. 5A), were tested in cell-based transfection experiments. As demonstrated in Fig. 5B, among all of the tested PBR ligands, only PK11195 substantially and picrotoxin moderately repressed the constitutive activity of hCAR, whereas PK11195 and FGIN-1-27 showed potent activations of hPXR, which were comparable with that of RIF, in reporter assays (Fig. 5C). A hCAR-splicing variant (hCAR3) has been identified with low basal activity, but it could be activated by CITCO in cell-based transfection assays (Auerbach et al., 2005; Faucette et al., 2007). Using an hCAR3 expression vector, our cell-based transfection assay revealed no obvious activation of hCAR3 by any of the tested PBR ligands (Fig. 5 D).
PK11195 Deactivates hCAR through a PBR-Independent Pathway. To further test whether the deactivation effect of PK11195 on hCAR is mediated through a PBR-signaling pathway, siRNA specific for PBR was used to knock down PBR expression in HeLa cells, which express high levels of endogenous PBR (Gonzalez-Polo et al., 2005). Forty-eight hours after transfection of siRNA specific for PBR, the expression of PBR gene in HeLa cells was significantly down-regulated compared with the control group transfected with nontargeting siRNA (Fig. 6A). However, the knockdown of PBR expression did not affect the deactivation effect of PK11195 on hCAR in parallel cotransfection assays (Fig. 6B). Thus, these results support the notion that PK11195-mediated inhibition of hCAR is PBR-independent.

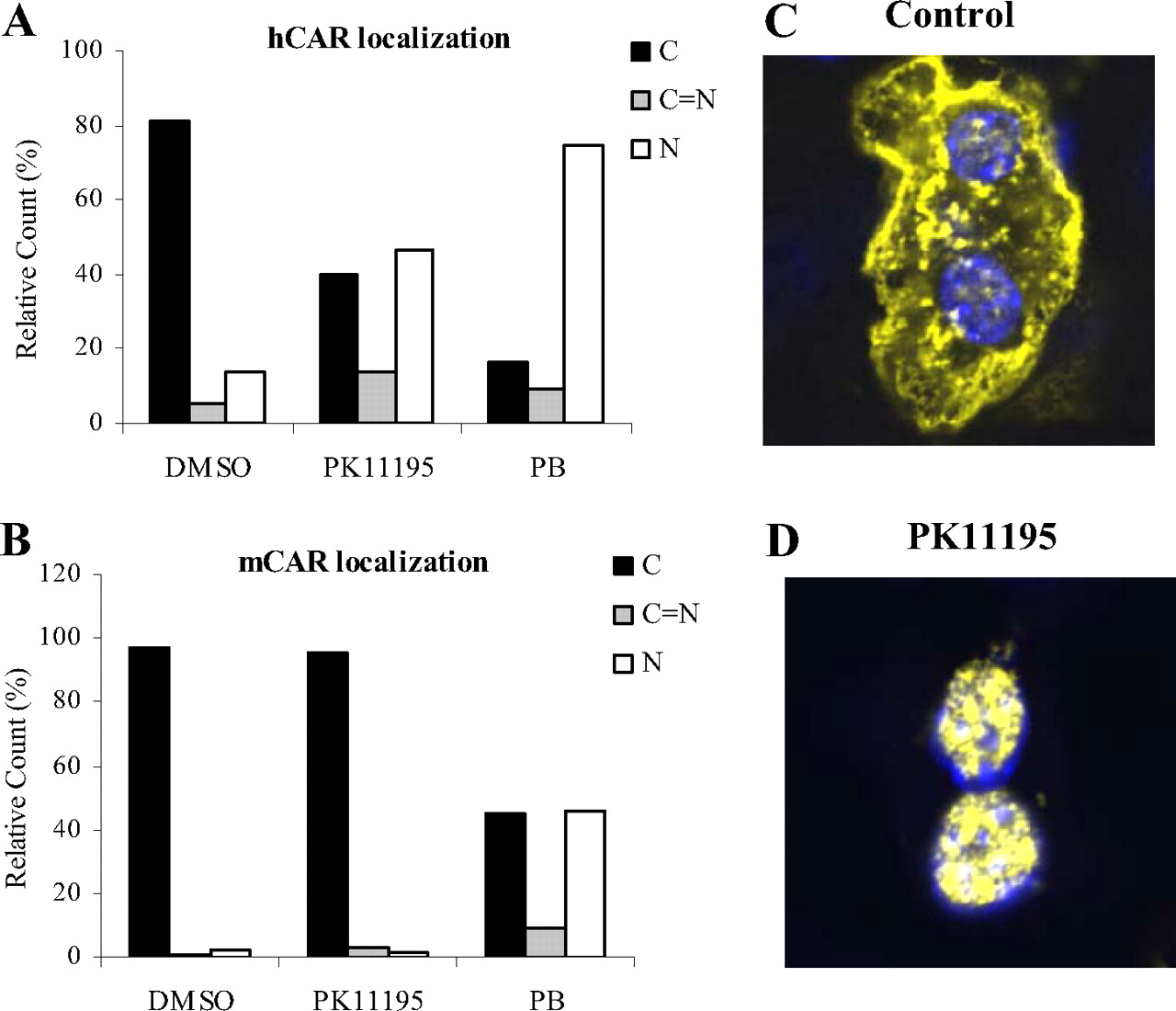
PK11195 translocates hCAR in CAR-/- mouse liver. Car-/- mice were injected with 10 μg of expression plasmid encoding EYFP-tagged hCAR or EYFP-mCAR as described under Materials and Methods and treated 3 h later with vehicle (DMSO, CT), 100 mg/kg PB, or 10 mg/kg PK11195. Mice livers were harvested 8 h after tail vein delivery, and slides of frozen liver sections were prepared for confocal microscopy. Approximately 100 EYFP-expressed cells were classified according to hCAR (A) or mCAR (B) localization status. Representative images depict examples of hCAR localization as EYFP-hCAR in yellow and nuclear staining in blue (C and D).
The Effect of PK11195 on CAR Translocation in CAR-/-Mice. CAR activation is a multistep process with nuclear translocation as the initial step. Our results thus far demonstrate that PK11195 deactivates hCAR in immortalized cell lines; therefore, evidence from a physiologically relevant system is required to establish whether this effect is exerted at CAR translocation or at CAR activation inside the nucleus. Car-/- mice were injected with EYFP-tagged hCAR or mCAR and treated with PK11195 or vehicle control as outlined under Materials and Methods. Confocal microscopic analysis of the liver sections revealed that EYFP-hCAR and FYFP-mCAR were primarily localized in the cytoplasm without treatment. Upon treatment with PK11195 or PB, EYFP-hCAR was efficiently accumulated in the nucleus (Fig. 7, C and D). Analysis of 100 hCAR-expressing cells in liver sections from each treatment group showed that hCAR nuclear localization increased from 13.6 (control) to 46.3 and 76%, whereas cytoplasmic localization decreased from 80 (control) to 40 and 17% after PK11195 and PB treatment, respectively (Fig. 7A). In contrast, the cellular distribution of EYFP-mCAR in the transfected liver has not been altered by the treatment of PK11195 (Fig. 7B). Overall, these results indicate that the deactivation of hCAR by PK11195 primarily occurred inside of the nucleus.
Interaction of hCAR with Coactivators Was Inhibited by PK11195. Because CAR was demonstrated to have constitutive interactions with several coactivators (Tzameli et al., 2000; Huang et al., 2004), mammalian two-hybrid and GST pull-down assays were used to further explore the mechanisms underlying the PK11195-mediated hCAR deactivation. In agreement with previous reports, hCAR was able to directly interact with SRC-1 and GRIP1 in the absence of ligand, and the addition of hCAR agonist CITCO further enhanced the interaction (Fig. 8A and B). It is noteworthy that both basal and CITCO-enhanced interactions of hCAR with SRC-1 and GRIP1 were significantly repressed by 10 μM PK11195. In our GST pull-down experiments, inclusion of CITCO clearly enhanced the direct interaction between hCAR and SRC-1 or GRIP1, and this increased binding was partially disrupted in the presence of PK11195 (Fig. 8E). Given that both recruitment of coactivators to and disruption of corepressors from NRs are accepted mechanisms associated with activation, we further tested whether PK11195 could recruit corepressors to hCAR. As shown in Fig. 8, C and D, two corepressors (NcoR and SMRT) only marginally bound to hCAR in the absence of ligand, and the addition of PK11195 did not increase these interactions. PXR constitutively interacted with NcoR and SMRT in the absence of ligand, and these interactions were partially disrupted by the presence of RIF.
Discussion
In this study, we have identified PK11195, a well known peripheral benzodiazepine receptor ligand used for the treatment of cancer, autoimmune diseases, and neurodegenerative diseases (Galiegue et al., 2003), is a selective and potent inhibitor of hCAR and effectively represses hCAR-mediated gene expression in different cell lines. Only two compounds, CLZ and meclizine, have been reported previously as selective hCAR antagonists with moderate deactivation activities. In cell-based transfection assays, the maximal deactivation of hCAR achieved by both CLZ and meclizine were approximately 50% of the constitutive activity levels in CV-1 and HepG2 cells (Tzameli et al., 2000; Huang et al., 2004). Moreover, CLZ's antagonistic effects on hCAR were also challenged by the observations that instead of repression, CLZ consistently activated hCAR in human embryonic kidney 293 cell transfection assays (Honkakoski et al., 2001) and activated a splicing variant of hCAR (hCAR3) in COS1 cells (Auerbach et al., 2005). In contrast, our results demonstrated that PK11195 (10 μM) could inhibit the activity of hCAR by 85% in HepG2 cells (Fig. 1B), and this potent inhibition of hCAR was also observed in cell-based reporter assays using several other cell lines, including human embryonic kidney 293 (data not shown). To our knowledge, PK11195 represents the most potent hCAR deactivator identified thus far.
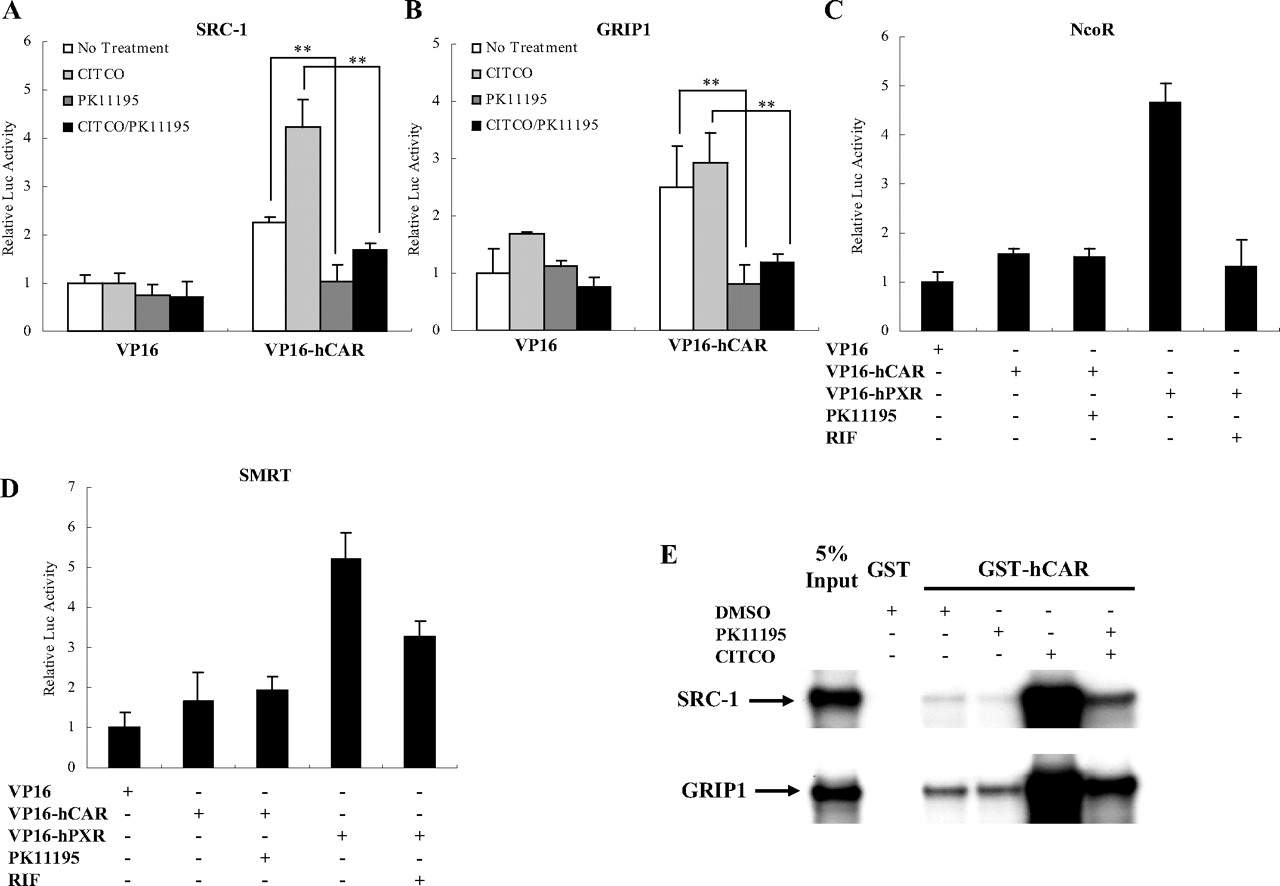
PK11195 disrupts coactivators binding to hCAR. Mammalian two-hybrid assays were performed in COS1 cells transiently transfected with expression plasmids encoding VP16-AD/hCAR fusion proteins and GAL4-DBD/coregulators fusion proteins, as indicated, together with the reporter gene plasmid pG5luc. Cells were treated with DMSO (0.1% v/v), CITCO (1 μM), or PK11195 (10 μM) for an additional 24 h before the determination of luciferase activities (A-D). Data represent the mean ± S.D. of three independent transfections (**, p < 0.01). E, GST pull-down assay. In vitro-translated 35S-labeled SRC-1 and GRIP-1 were incubated with bacterially expressed GST-hCAR fusion protein in the presence of 0.1% DMSO, 100 μM PK11195, or 1 μM CITCO as indicated. GST was used as a negative control for binding; in vitro incubation, gel electrophoresis, and autoradiography were done as described under Materials and Methods.
Recent studies established that differences between hCAR and their rodent counterparts dictate the species-specific expression of target genes. For example, TCPOBOP activates mouse but not human CAR, androstanol represses mouse but not human CAR, and meclizine represses human but activates mouse CAR. Here, we show that PK11195 exhibited negligible effects on mouse CAR activation in multiple transfection assays. It is interesting that PK11195 also displayed potent activation of hPXR, complicating its effect on several PXR and CAR shared target genes. Indeed, PK11195 induced the expression of both CYP2B6 and CYP3A4 in human primary hepatocytes in which endogenous expression of NRs are well maintained. Furthermore, in agreement with in vitro reporter assays, the observed inductions of CYP2B6 and CYP3A4 in human hepatocytes were significantly repressed by a selective hPXR deactivator SFN. Given that human CAR and PXR hold both common and distinct target genes, PK11195 would provide a valuable chemical tool to illustrate CAR-specific roles in regulating gluconeogenesis and fatty acid oxidation (Sugatani et al., 2001; Kodama et al., 2004; Ueda et al., 2005). In addition, Car-/- mice displayed enhanced weight loss during caloric restriction, suggesting that PK11195 may represent a potentially novel drug target for uncoupling metabolic rate from food intake with implications to obesity and its associated disorders (Maglich et al., 2004; Huang et al., 2005).
CAR displays unique activation mechanisms compared with other orphan nuclear receptors, requiring both nuclear translocation and nuclear activation. It is noteworthy that nuclear translocation of CAR could be induced by both indirect activators such as PB-type compounds and agonistic ligands such as CITCO. Several known mCAR deactivators exhibit their effects through blocking the nuclear accumulation of mCAR. For example, pretreatment of primary mouse and rat hepatocytes with okadaic acid, a protein phosphatase 2A inhibitor, results in decreased CAR nuclear accumulation and CYP2B induction by PB and TCPOBOP (Honkakoski and Negishi, 1998; Kawamoto et al., 1999). The mouse antagonists, androstanes, also cause CAR cytoplasmic retention in mouse hepatocyte cultures. In contrast, our results in CAR-null mice showed that PK11195 efficiently induced nuclear translocation of transfected EYFP-hCAR, even though it deactivated the constitutive activity of hCAR in HepG2 cells in which hCAR was spontaneously accumulated in nuclei. It was also reported previously that TCPOBOP induced translocation of human CAR in CAR-null mice but was incapable of activating a Cyp2b10-PBREM reporter gene (Honkakoski et al., 2003). Together, these results argue against constitutive activation of nuclear localized CAR and indicate that nuclear activation is one of the two required steps for target gene activation.
Several coactivators and corepressors have been reported to modulate the function of nuclear localized CAR. Ligand-independent interaction of hCAR with SRC-1, PGC-1, and GRIP-1 has been purported to confer constitutive activity to CAR (Muangmoonchai et al., 2001; Shiraki et al., 2003; Miao et al., 2006). Present studies further demonstrated that PK11195 could effectively inhibit coactivators (SRC-1 and GRIP1) binding to hCAR in mammalian two-hybrid and GST pull-down assays. In contrast, the low binding of hCAR with corepressors (NcoR and SMRT) was not changed after the treatment of PK11195. Because coactivator assembly was necessary for hCAR function in the nucleus (Tzameli et al., 2000; Huang et al., 2004), these observations suggest that PK11195 mediates the inhibition of hCAR activity by disrupting the interaction of coactivators with hCAR in nucleus.
PK11195 is a typical ligand of the peripheral benzodiazepine receptor, which is located at the surface of the mitochondria and has been associated with numerous biological functions, such as regulation of cell proliferation, stimulation of steroidogenesis, immunomodulation, and regulation of mitochondrial functions (Galiegue et al., 2003). Several structurally unrelated ligands of PBR, including picrotoxin, Ro5-4864, FGIN-1-27, and CMZ, have been tested in current studies to evaluate their potential roles on hCAR activity. It is intriguing that PK11195 is the only PBR ligand that strongly repressed hCAR activation in cell-based transfection assays. Further investigation showed that PK11195 deactivation of hCAR activity was not affected by knockdown expression of the endogenous PBR gene. Therefore, these results indicate that the antagonistic effect of PK11195 on hCAR is mediated through PBR-independent signaling pathways. Because PK11195 exerts promising clinical applications, including regulation of cholesterol transport, synthesis of steroid hormones, and potential cancer therapeutics by enhancing apoptosis (Gonzalez-Polo et al., 2005), our present study could potentially broaden the therapeutic scope of PK11195.
In conclusion, accumulated evidence suggests that CAR may have evolved in mammals to function as both a metabolic “stress” receptor and a xenobiotic receptor. Despite its involvement in several physiologically protective mechanisms, CAR also may contribute to liver toxicity by exerting the same effects usually considered beneficial. Moore and colleagues demonstrated that CAR enhances acetaminophen toxicity by inducing enzymes involved in toxic metabolite formation and glutathione depletion (Zhang et al., 2002). Furthermore, CAR activation by PB and TCPOBOP results in sensitization to cocaine hepatotoxicity (Wei et al., 2000). Finally, CAR activation has been shown to mediate PB-induced liver tumor promotion in mice (Yamamoto et al., 2004; Huang et al., 2005). Thus, there seems to be a tenuous balance between the positive and negative effects of CAR. Because these beneficial and detrimental physiological effects of CAR have been demonstrated primarily with rodent isoforms, identification of PK11195 as a selective and potent hCAR deactivator will provide a unique tool for obtaining additional insight into the mechanism underlying hCAR activation.
Acknowledgments
We thank Rick Moore (National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC) for helping with the mouse tail vein injection assays and Drs. Steve Kliewer (University of Texas, Southwestern Medical Center, Dallas, TX), David Moore (Baylor College of Medicine, Houston, TX), and Bryan Goodwin (GlaxoSmithKline, Research Triangle Park, NC) for kindly providing pSG5-hPXR, GAL4-NCoR expression vectors and CYP3A4-PXRE/XREM reporter vector, respectively. We also thank Dr. Leslie Tompkins for insightful comments and critical reading of the manuscript. Human liver tissues were procured with the assistance of John Cottrell from the University of Maryland Baltimore Medical Center (Baltimore, MD).
Footnotes
-
This research was supported by National Institutes of Health grant DK061652 (to H.W.) and by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
-
ABBREVIATIONS: CAR, constitutive androstane receptor; PXR, pregnane X receptor; PBR, peripheral benzodiazepine receptor; PK11195, 1-(2-chlorophenyl-N-methylpropyl)-3-isoquinoline-carboxamide; NR, nuclear receptor; PB, phenobarbital; CITCO, 6-(4-chlorophenyl)imidazo-[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl) oxime; RIF, rifampicin; CMZ, carbamezapine; CLZ, clotrimazole; SFN, sulforaphane; SRC-1, steroid receptor coactivator-1; GRIP-1, glucocorticoid receptor-interacting protein 1; NcoR, nuclear receptor corepressor; SMRT, silencing mediator of retinoid and thyroid hormone receptor; EYFP, enhanced yellow fluorescent protein; siRNA, small interfering RNA; PBREM, phenobarbital-responsive enhancer module; PXRE, pregnane X receptor response element; XREM, xenobiotic-responsive enhancer module; kb, kilobase(s); DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; MOPS, 4-morpholinepropanesulfonic acid; CT, vehicle control; TCPOBOP, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene; FGIN-1-27, N,N-dihexyl-2-(4-fluorophenyl)-indole-3-acetamide; Ro5-4864, 7-chloro-5-(4-chlorophenyl)-1,3-dihydro-1-methyl-2H-1,4-benzodiazepine-2-one.
- Received February 22, 2008.
- Accepted May 20, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}