


Abstract
By extending previous studies of the phenobarbital (PB)-responsive 132-base pair (bp) enhancer sequence in the CYP2B10gene, we have delimited a 51-bp enhancer element that is fully inducible by PB in mouse primary hepatocytes. Sixteen structurally unrelated phenobarbital-type inducers activated the 51-bp enhancer element in transient transfection assays. The results thus indicate that most PB-type inducers, if not all inducers, increase the transcription of the CYP2B10 gene by activating this 51-bp element, now designated PB-responsive enhancer module or PBREM.
The induction of P450s and other drug-metabolizing enzymes by xenobiotic chemicals is a common cellular defense mechanism against the toxicity and carcinogenicity of foreign compounds. Inducers can be classified into different groups depending on the subset of P450 genes they activate (Okey, 1990). One of the two major groups, polycyclic aromatic hydrocarbons and dioxins, consists of xenochemicals that activate the xenobiotic response element of CYP1A genes via the aryl hydrocarbon receptor-mediated mechanism (Hankinson, 1995; Whitlocket al., 1996). The other major group of P450 inducers, typified by PB, consists of a host of structurally diverse xenochemicals that induces a subset of P450 genes within theCYP2A, 2B, 2C, and 3Asubfamilies, with the CYP2B genes being most effectively up-regulated (Okey, 1990; Lubet et al., 1992; Waxman and Azaroff, 1992; Nims and Lubet, 1995; Whitlock et al., 1996;Honkakoski and Negishi, 1998).
Working independently, three laboratories have recently associated the PB-responsive enhancer activity with the function of DNA sequences at −2.4 to −2.3 kbp in distal regions of the rat CYP2B2 and mouse CYP2B10 genes, respectively (Trottier et al., 1995; Park and Kemper, 1996; Honkakoski and Negishi, 1997). The 132-bp mouse sequence PBREM conferred an 8- to 10-fold induction by PB and TCPOBOP to a heterologous TK promoter in mouse primary hepatocytes (Honkakoski and Negishi, 1997). Within the PBREM, we have subsequently identified the 33-bp core element that contains NR-binding half sites and an NFI site. This 33-bp core element, however, displayed only about 3-fold induction of the enhancer activity, indicating that the full enhancer activity of PBREM required sequences outside the 33-bp core (Honkakoski and Negishi, 1997). We have defined here a fully PB-inducible 51-bp enhancer element that includes the previous 33-bp core. We have also found that this 51-bp element responds to numerous known PB-type inducers in mouse primary hepatocytes.
Experimental Procedures
Reagents.
[α-32P]dATP (>6000 Ci/mmol) and [14C]dichloroacetylchloramphenicol (56 mCi/mmol) were purchased from Amersham (Arlington Heights, IL). High purity pesticides and chlorinated biphenyls were obtained from Accustandard (New Haven, CT) and high performance liquid chromatography-grade solvents were from Sigma-Aldrich (St. Louis, MO). All other chemicals and enzymes were from Boehringer Mannheim (Indianapolis, IN), Sigma Chemical (St. Louis, MO), or GIBCO BRL (Gaithersburg, MD).
Plasmids.
The 177-bp CYP2B10 DNA fragment (−2426/−2250 bp) containing the PB-responsive PBREM enhancer has been described previously (Honkakoski and Negishi, 1997). Various 5′ and 3′ deletion constructs were generated using appropriate 20–24-mer primers harboring a BamHI site at the 5′-end for cloning purposes (Fig. 1). The amplified DNAs for these deletion constructs were digested with BamHI and ligated into BamHI site of pBLCAT2 (TKCAT) plasmid (Luckow and Schütz, 1987). Nucleotide mutation of the 51-bp enhancer element (−2339/−2289 bp) was done in similar fashion, using XbaI and BamHI sites of TKCAT plasmid as the 5′ and 3′ cloning sites, respectively [NR1a2a mutant (both NR1a and NR2a were mutated to TCTGGT) and NR1b2b mutant (both NR1b and NR2b to TCTGGT)]. Appropriate recombinant plasmid DNAs produced in Escherichia coli TG-1 cells were purified twice on CsCl gradients and verified by DNA sequencing over the amplified regions. The quality and supercoiling of plasmid DNAs were checked by agarose gel electrophoresis. The indicated folds of the induction were averaged from the two independent cell batches.

A, Deletion assays to delimit the 51-bp enhancer element. The transfected cells were cultured for 24 hr in the presence of DMSO vehicle (−) or 50 nm TCPOBOP (+) before CAT assays. The fold inductions shown are means from two to three transfections using the independent cell batches. B, The DNA sequences corresponding to the 51-bp enhancer element from the PB-inducible mouse CYP2B10 (GenBank accession no. U67059), rat CYP2B1 (U30327) and CYP2B2 (S51970), and noninducible mouse CYP2B9 (M60267).Boxed, the two putative nuclear receptor binding motifs (designated NR1 and NR2 with a and b for their half sites) and the NFI binding sequence; horizontal lines on theCYP2B10 gene, pC footprint (Honkakoski and Negishi, 1997).
Transient transfection of mouse primary hepatocytes.
Two-month-old C57BL/KS/J male mice were purchased from Jackson Laboratory (Bar Harbor, MA). About 15 × 106mouse hepatocytes isolated by two-step collagenase perfusion (Honkakoski et al., 1996) were electroporated with 30 μg each of individual enhancer-TKCAT reporter plasmids and 10 μg of pSVβgal control plasmid (Promega, Madison, WI) to normalize results between different plasmid DNAs as described previously (Honkakoski and Negishi, 1997). Transfected cells on dishes (2–3 × 106 cells) were cultured for 24 hr in the absence or presence of inducers under the conditions previously described (Honkakoski and Negishi, 1997). The chemicals used and their concentrations were: drugs, 1 mm PB, 1 μmclotrimazole, 8 μm chlorpromazine, or 200 μm metyrapone; solvents, 12 mm acetone, 2 mm methyl isobutyl ketone, 8 mm isoamyl alcohol, or 2 mm pyridine; PCBs, 5 μm2,2′,4,4′-tetrachlorobiphenyl, 5 μm2,2′,5,5′-tetrachlorobiphenyl, or 5 μm2,3,3′,4′,5,6-hexachlorobiphenyl; pesticides, 10 μmdieldrin, 10 μm1,1,1-trichloro-1,2-bis(o,p′-chlorophenyl)ethane, or 10 μm methoxychlor; plant product, 200 μmcamphor. For the positive and negative controls, the transfected cells were treated with 50 nm TCPOBOP and 50 nm1,4-bis[2-(3-chloro-pyridyloxy)]benzene (an inactive derivative of the potent inducer TCPOBOP), respectively. Five micromoles/liter 3-methylcholanthrene (CYP1A1 inducer) was employed to examine the enhancer specificity to the PB-type inducers. These chemicals were dissolved in DMSO, saline or ethanol. Cell extracts (Pothier et al., 1992) were assayed for protein (Bradford, 1976) or β-galactosidase (Alam and Cook, 1990), heat-treated for 20 min, and assayed for CAT activity (Hattori et al., 1990). In addition, RNAs were extracted from the transfected hepatocytes for Northern analysis of the endogenous CYP2B10 and albumin mRNAs (Honkakoski et al., 1996).
Results
The 51-bp sequence as the functional enhancer element.
Detailed deletions of the 132-bp PBREM (position −2397/−2265 bp) were designed so as to extend the characterization of the 33-bp core without limiting it to the locations of the previous DNase I footprints (Honkakoski and Negishi, 1997), as shown in Fig. 1A. The 3′ deletions from −2265 bp to −2288 bp did not affect the PB-inducibility of the PBREM (10–11-fold; Fig. 1A, lanes 5–10), whereas successive deletion of the −2307/−2288 bp fragment decreased the induction to 2.8-fold (Fig. 1A, lanes 11 and 12). Further deletion of the −2333/−2306 bp fragment showed a complete loss of induction (lanes 13–14). On the 5′ deletions, removal of DNA up to −2340 bp had no major effect on the PB-induced CAT activity (Fig. 1A, lanes 15–20). Further deletion of the −2339/−2334 bp fragment decreased the induction to 4.5-fold (Fig. 1A,lanes 21–22). When the 3′- and 5′-deletions were taken into account in selecting a minimal enhancer element, the 51-bp fragment (−2339/−2289 bp) exhibited the same 11-fold induction as the 132-bp PBREM (Fig. 1A, lanes 23–24).
Sequence analysis of the 51-bp enhancer element revealed two apparent binding sites for members of the NR family and an NFI binding site (Fig. 1B). These NR binding sites were composed of imperfect direct repeats of AGGTCA-like half sites with a spacing of four nucleotides (DR4 motif), designated here as NR1 (TGTACTttccTGACCT at −2337 bp on the top strand) and NR2 (TCAACTtgccTGACAC at −2305 bp on the top strand) (Fig. 1B); their 5′- and 3′-half sites were named a and b, respectively. In addition to the perfect half site NR1b, our previously identified 33-bp core covered the NFI and NR2a (Fig. 1B,horizontal lines). The NR1a was located between the pB′ and pC regions described previously (Honkakoski and Negishi, 1997), whereas the NR2b was found in the sequences between the pC and pD regions. Sequence comparisons of the 51-bp element with the corresponding sequence of the PB-noninducible mouse CYP2B9 gene revealed many nucleotide mutations within NR1a, NR1b, a core repeat of the NFI site, and NR2b. Our previous studies have shown that these mutations abolish the PB inducibility of PBREM (Honkakoski and Negishi, 1997). The PB-inducible rat CYP2B genes, on the other hand, had no mutation within these critical repeats, although they had a single nucleotide mutation and/or insertion within the spacing regions of the NR sites.
Response of the 51-bp element to diverse chemicals.
A prominent feature of CYP2B gene induction is its responsiveness to structurally diverse xenochemicals (Lubet et al., 1992; Waxman and Azaroff, 1992; Nims and Lubet, 1995;Honkakoski and Negishi, 1998). Thus, we wanted to see whether the 51-bp enhancer element could be activated in response to these xenochemicals. For this purpose, in addition to PB and TCPOBOP (Fig 2A, lanes 2 and 15), sixteen xenochemicals were selected based on their reported ability to induce the CYP2B genes in rodents and were examined for their capability to activate the 51-bp element (Fig. 2A): drugs (lanes 2–4), organic solvents (lanes 7–10), PCBs (lanes 12–14), pesticides (lanes 17–19), and plant products (lane 20). First of all, Northern hybridization of RNAs extracted from the same transfected hepatocytes confirmed that the endogenous CYP2B10 mRNAs were in fact induced by these 16 known PB-type xenochemicals (Fig. 2A). Subsequently, these primary hepatocytes transfected with a CAT reporter gene under the control of the 51-bp enhancer element were examined for inducibility by these individual xenochemicals (Fig. 2B). This resulted in 3.1 ± 0.2-fold (by isoamyl alcohol) to 11.5 ± 2.7-fold (by TCPOBOP) increases in CAT activity, which indicated that the enhancer element responded to all of these xenochemicals. The inducibilities of the 51-bp enhancer-dependent CAT activity correlated well with the induced levels of the CYP2B10 mRNA. As expected, the CYP1A inducer 3-methylcholanthrene (Fig 2B,lane 23), the inactive derivative 3, 3′-DCP (Fig 2B,lane 24), and the CYP2E inducer enthanol (Fig 2B, lane 22) were not able to activate the 51-bp element. To examine whether the activation of the 51-bp enhancer element was sequence specific, we mutated the NR half sites and transfected these mutated 51-bp enhancer TKCAT plasmids into primary hepatocytes. The mutant NR1a2a were not able to respond to either PB (Fig. 2C, lane 1) or TCPOBOP (Fig. 2C, lane 15). Similarly, other mutant NR1b2b also failed to respond to these inducers (data not shown). The importance of the half sites for the induction response confirmed our previous finding with the naturally mutated, noninducibleCYP2B9 gene (Honkakoski and Negishi, 1997). These results provide compelling evidence that the induction by many “PB-like” inducers, if not all, is mediated through the 51-bp enhancer element.
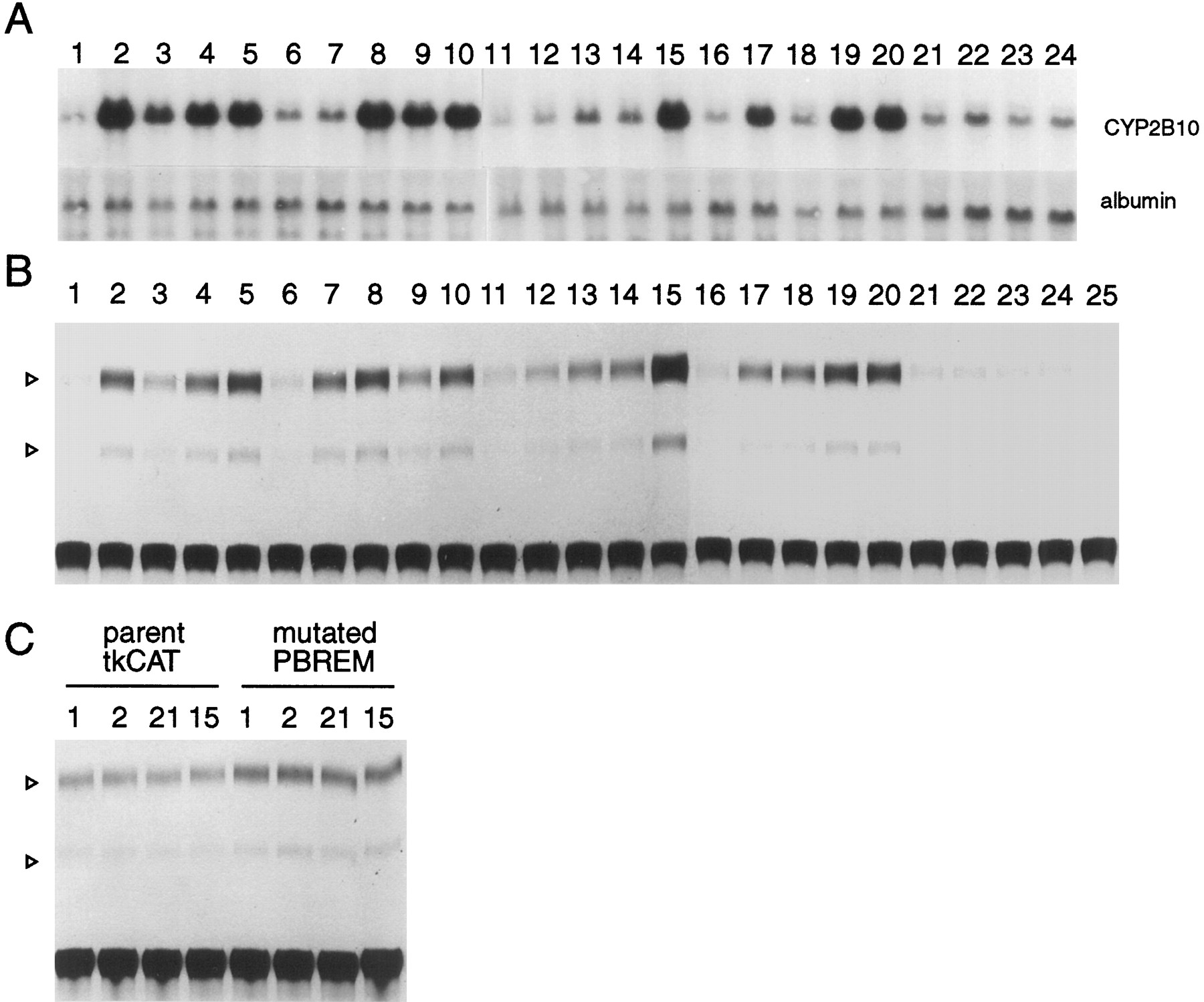
RNA and cell extract were prepared from the transfected cells that were treated with different inducers (except B,lane 25, in which extract from untransfected cells was used in the CAT assays). A, Cells subjected to Northern hybridization. B, Cells subjected to CAT assays. Lanes correspond to the same inducers in different parts of the figure: lane 1, DMSO; lane 2, PB (9.8 ± 2.8);lane 3, clotrimazole (3.3 ± 1.0); lane 4, chlorpromazine (5.9 ± 1.1); lane 5, metyrapone (7.5 ± 1.7); lane 6, DMSO; lane 7, acetone (4.2 ± 0.5); lane 8, methyl isobutyl ketone (9.5 ± 1.5); lane 9, isoamyl alcohol (3.1 ± 0.2); lane 10, pyridine (5.4 ± 1.2); lane 11, DMSO; lane 12, 2,2′,4,4′-tetrachlorobiphenyl (3.3 ± 0.4); lane 13, 2,2′,5,5′-tetrachlorobiphenyl (4.1 ± 0.3);lane 14, 2,3,3′,4′,5,6-hexachlorobiphenyl (5.0 ± 1.1); lane 15, TCPOBOP (11.5 ± 2.7); lane 16, DMSO; lane 17, dieldrin (5.2 ± 1.2); lane 18, 1,1,1-trichloro-1,2-bis(o,p′-chlorophenyl)ethane (4.8 ± 0.8); lane 19, methoxychlor (7.3 ± 0.6); lane 20, camphor (6.7 ± 1.1); lane 21, saline (1.1 ± 0.2); lane 22, ethanol (1.0 ± 0.1); lane 23, 3-methylcholanthrene (1.0 ± 0.2); lane 24, 1,4-bis[2-(3-chloro-pyridyloxy)]benzene (1.0 ± 0.1).Lane 25, extract from untransfected cells used in CAT assay. C, Hepatocytes were transfected with control TKCAT (tkCAT) or with mutated PBREM-tkCAT plasmids. The numbers correspond to those in B.
Discussion
Induction of hepatic microsomal drug metabolism by barbiturates was first reported 35 years ago (Remmer and Merker, 1963). Since then, the induction has long been implicated as an important factor for the pharmacological, toxicological, and carcinogenic effects of xenochemicals (Conney, 1967; Conney, 1982). A transgenic mouse study was the first to indicate that proximal sequences of CYP2B gene are not sufficient to regulate the PB-responsive transcription and that the PB-responsive element may be located in the far distal region of the genes (Ramsden et al., 1993). The mechanism by which PB and PB-type inducers regulate the P450 genes, however, remained elusive until a 177-bp DNA element at −2318/−2155 of the rat gene was reported to contain a functional PB-responsive enhancer activity (Trottier et al., 1995). Subsequently, two other laboratories have confirmed independently that the PB-responsive enhancer activity resides within the corresponding DNA sequences in the rat as well as mouse CYP2B genes (Park and Kemper, 1996;Honkakoski and Negishi, 1997). Moreover, an in vivofootprinting assay has recently shown a PB-responsive alteration in binding of nuclear protein to the enhancer sequence (Kim and Kemper, 1997). Our present studies with the mouse CYP2B10 gene have now identified the 51-bp DNA sequence as the general enhancer element that responds to diverse xenochemicals to regulate PB-inducible transcription of the CYP2B genes. We now call this 51-bp enhancer sequence PBREM, instead of the 132-bp DNA designated previously (Honkakoski and Negishi, 1997).
The 51-bp PBREM seems to be a composite enhancer element that contains multiple nuclear protein binding sites [(orphan) NRs and NFI]. The repeat sequences of these binding sites are 100% conserved in the PB-inducible mouse and rat CYP2B genes, whereas they are mutated in the PB-nonresponsive mouse CYP2B9 gene. It has been shown that these nucleotide mutations within NR sites of theCYP2B9 gene abolish the PB-inducibility of the PBREM activity (Honkakoski and Negishi, 1997). Because nuclear receptors are far more diverse in their structure and function than is NFI, we proposed previously that the perfect NR half site of the 132-bp enhancer element might be a primary target of the PB signal (Honkakoski and Negishi, 1997; 1998). A sequence analysis of the 51-bp PBREM has now suggested that this half site (NR1b) is a part of a putative NR binding site NRI. The binding of an NR to its site is known to be dictated by the sequence, the orientation, and the spacing of the half sites (Mangelsdorf et al., 1995). If NRI is, in fact, an NR binding site with a DR4 motif of imperfect repeats, an NR heterodimer may be a possible candidate for binding to the NRI site. Once these proteins have been identified (in future studies), we should be able to find the regulatory mechanism of the PBREM activity in response to PB-type inducers.
Although certain PB-like inducers such as PCB congeners display some structure-induction relationship, numerous xenochemicals with distinct structures (e.g., pyridine versus methoxychlor) can activate the 51-bp PBREM. This fact implies that PB-like inducers may not have a common target until their signals converge on the PBREM. This induction mechanism would distinguish it from the direct aryl hydrocarbon receptor binding by structurally similar polycyclic aromatic hydrocarbon and dioxin ligands to activate the cognate xenobiotic response element of the CYP1A genes (Hankinson, 1995;Whitlock et al., 1996). Because the 51-bp PBREM contains the nuclear receptor binding sites, one possibility is that various chemicals act through the PBREM via multiple pathways such as activation of DNA-binding factors by direct ligand binding or by signal transduction through phosphorylation, as described for nuclear receptors including estrogen receptor and retinoid X receptors (Leidet al., 1992; Bunone et al., 1996). Alternatively, different nuclear receptor heterocomplexes, such as the retinoid X receptor, can be activated and can bind to PBREM, depending upon the exposure to different PB-type inducers. Orphan receptors such as the retinoid X receptor and the chicken ovalbumin upstream promoter transcription factor can be ideal for this role because they can form heterocomplexes with other nuclear (orphan) receptors (Mangelsdorf and Evans, 1995). Additionally, it still remains to seen whether a protein binds to diverse xenochemicals and transduces their inducing signals to the PBREM. It should not be ruled out, however, that the PB induction can be also regulated by factors aside from PBREM activation. Some inducers (e.g., acetone, etc.) exhibited less correlation between the induction of CAT activity and the induction of CYP2B10 mRNA, implying that other factors such as mRNA stability may also be involved in the PB induction, depending on the types of inducers. Regardless of which of the above possibilities explains the PB-induction mechanism, the identification of the 51-bp PBREM should accelerate the research on the molecular mechanism of PB-inducible transcription of the CYP2B genes and tens of other genes known to be induced by PB (Frueh et al., 1997).
The central role of the 51-bp PBREM for CYP2B gene induction has wide implications, not only in the field of gene regulation, but also in the understanding of the toxicological effects of some persistent environmental contaminants and widely used drugs, helping to define occupational health hazards. Moreover, the responsiveness of the PBREM to diverse xenochemicals may lead to some practical applications, such as a drug-regulated enhancer in gene therapy, a cell culture model, or a cell culture- or transgenic animal-based test system for identifying toxic chemicals through their ability to induce the P450 genes.
Acknowledgments
We thank Drs. Igor Zelko and Cary Weinberger for their helpful discussion and comments on this manuscript.
Footnotes
- Received December 12, 1997.
- Accepted January 9, 1998.
-
Send reprint requests to: Dr. Masahiko Negishi, Pharmacogenetics Section, Lab of Reproductive and Developmental Toxicology, NIEHS, NIH, Research Triangle Park, NC 27709. E-mail:negishi{at}niehs.nih.gov
-
↵1 Current affiliation: Department of Pharmaceutics, University of Kuopio, FIN-70211 Kuopio, Finland.
Abbreviations
- P450
- cytochrome P450
- bp
- base pairs
- CAT
- chloramphenicol acetyltransferase
- DMSO
- dimethyl sulfoxide
- NFI
- nuclear factor I
- NR
- nuclear receptor
- PB
- phenobarbital
- PBREM
- phenobarbital-responsive enhancer module
- PCB
- polychlorinated biphenyl
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- TK
- thymidine kinase
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}