


Abstract
Previous analyses suggested that potent aryl hydrocarbon receptor (AhR) antagonists were planar, with a lateral electron-rich center. To further define structural requirements and mechanism for antagonism, ten additional flavone derivatives were synthesized. Based on their ability to 1) compete with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) for binding to the AhR; 2) inhibit TCDD-elicited binding of AhR to dioxin-responsive elements (DRE) in vitro; and 3) inhibit TCDD-induced transcription of DRE-dependent luciferase in stably transfected hepatoma cells, the most potent flavones contained a 3′-methoxy group and a 4′-substituent having one or more terminal atoms of high electron density (−N3, −NO2, or −NCS). Furthermore, these had low agonist activity as assessed by their inability to elicit AhR · DRE binding or to induce luciferase. Compounds containing bulkier 3′ or 4′-substituents, or a 3′-OH group were less potent antagonists, and some were partial agonists. In rat liver cytosol, 3′-methoxy-4′-azido- and 3′-methoxy-4′-nitroflavones bound competitively (with TCDD) to the AhR, indicating that they bind to the TCDD-binding site. When hepatoma cells were exposed to these flavones, AhR complexes were primarily immunoprecipitable from the cytosol and contained 90 kDa heat shock protein. In contrast, AhR in TCDD-treated cells was primarily immunoprecipitated from nuclear extracts and was associated with Arnt but not 90 kDa heat shock protein. Immunocytofluorescence analysis in intact cells further indicated that the potent antagonist inhibited nuclear uptake of AhR and blocked TCDD-dependent down-regulation of AhR. Together, these data indicate that the most potent antagonists bind the AhR with high affinity but cannot initiate receptor transformation and nuclear localization.
The binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related halogenated aromatic hydrocarbons to the aryl hydrocarbon receptor (AhR) is the first and key step in a series of molecular events leading to the binding of this transcription factor tocis-acting dioxin-responsive elements (DRE) and to the modulation of gene expression (reviewed in Whitlock, 1990; Hankinson, 1995). It has been postulated that the inappropriate and/or prolonged induction/repression of genes regulated by the AhR affects cellular differentiation states, and this leads to the numerous and species-specific toxic responses observed after exposure to these chemicals. The biochemical and functional characteristics of the AhR and the types of biological effects elicited after exposure to TCDD suggest that this protein may have some physiological function regulated by an endogenous ligand. Studies in AhR-deficient mice suggest a role in the development of the liver and immune system (Fernandez-Salguero et al. 1995; Schmidt et al. 1996). However, neither an endogenous ligand for the AhR nor its normal biological functions have yet been elucidated.
Because of the potent ability of TCDD and structurally-related chemicals to elicit the induction of a variety of genes, particularly those involved in drug metabolism and cellular growth processes (Bock, 1993), there has been considerable interest in identifying other ligands that bind to the AhR and elicit similar responses. Most of those identified to date are chemicals such as the chlorinated dioxins, dibenzofurans, azobenzenes, and certain biphenyls, as well as polycyclic aromatic hydrocarbons used in or resulting from a variety of industrial processes (Safe, 1990). More recently, chemicals found in foods or derived from naturally occurring compounds, and some of potential therapeutic use, have also been recognized as binding to the AhR. These chemicals include a variety of indoles (Gillner et al. 1985;Fernandez et al. 1988; Bjeldanes et al. 1991), benzocoumarins (Liu et al. 1993), substituted flavonoids (Lu et al. 1996; Ciolino et al. 1998), tryptanthrins (Schrenk et al. 1997), and photooxidized products of tryptophan (Rannug et al. 1987). The rank order of AhR binding affinity for these chemicals appears to be largely dependent on those structural constraints previously described (Gillner et al. 1993;Waller and McKinney, 1995): planar aromatic compounds, with approximate van der Waals dimensions of 14 × 12 × 5 Å, and with few bulky substituent groups. Several of these AhR ligands, however, act as partial antagonists under a variety of in vitro and in vivo conditions (Harris et al. 1989; Mahon and Gasiewicz, 1992;Kurl et al. 1993; Liu et al. 1993; Lu et al. 1996; Gasiewicz et al. 1996). Other than conforming to the structural features necessary for AhR binding, there are no obvious structural similarities among these compounds that would suggest requirements for antagonist activity.
Analysis of a broad range of substituted ellipticines and flavones suggests that the presence of an electron-rich center near or along a lateral position of the molecule as it fits within the van der Waals binding cavity of the AhR might be a characteristic that enhances antagonist activity (Gasiewicz et al. 1996). Although the substituted flavones appeared to be more potent (Lu et al. 1995; Gasiewicz et al. 1996), the small number of these structures examined limited identification of the important substituents necessary for antagonist activity. To further test, strengthen, and more precisely define our initial tentative hypothesis, a number of additional flavone derivatives were designed and synthesized for the current studies. Qualitative and quantitative analyses of these structures confirm and expand this hypothesis and suggest a molecular basis for the antagonist activity of these ligands. Furthermore, we show that, when the AhR is bound to the most potent of these flavones, it remains in the cytosol, associated with hsp90, and consequently fails to initiate the down-regulation of the AhR that is observed in TCDD-exposed cells. Together, the results suggest that the conformational change that initiates ligand-induced AhR transformation to a transcriptionally active form is dependent on structural features of the ligand in addition to those necessary for binding.
Materials and Methods
Chemicals.
The flavone compounds were synthesized by the procedure of Cunningham et al. (1992). Structures and purities (>98%) were confirmed by 1H-NMR spectroscopy, thin-layer chromatography, determination of melting point, and, for key compounds, combustion analysis. [3H]TCDD was purchased from Chemsyn Science Laboratories (Lenexa, KS), and unlabelled TCDD was purchased from Cambridge Isotopes (Cambridge, MA). [γ-32P]-ATP was purchased from NEN Research Products (Boston, MA). Oligonucleotides used for electrophoretic mobility shift assay (EMSA) were synthesized by Biosynthesis (Lewisville, TX).
Animals and Cytosol Preparation.
Male Sprague-Dawley rats (250–300 g; Charles River, Wilmington, MA) were maintained on a 12-h light cycle with ad libitum food and water. Rats were sacrificed by CO2 overdose, and livers were perfused and homogenized as described previously (Gasiewicz and Bauman, 1987) using HEDG buffer (25 mM HEPES, 1.5 mM Na2EDTA, 1 mM dithiothreitol, 10% (v/v) glycerol, pH 7.6 adjusted at room temperature). Protein concentration was measured by the method ofWaddell (1956), adjusted to approximately 15 mg/ml, and aliquots were stored at −70° until used.
Cell Culture and Preparation of Cytosols and Nuclear Extracts.
Mouse hepatoma cells, Hepa 1c1c7, were grown at 5% CO2 in modified Eagle’s medium (Sigma, St. Louis, Mo) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY), sodium pyruvate, l-glutamine, sodium bicarbonate, and Gentamicin. Ligands were administered in dimethylsulfoxide (1 μl/ml medium) to cultures when they were at least 90% confluent. Cells were harvested after 1 h and homogenized in HEDG buffer containing 0.4 μM leupeptin, 4 μg/ml aprotinin, 0.3 mM phenylmethylsulfonyl fluoride. For nuclear extracts, the pellet from a 20-min spin at 1000g was washed twice with the above buffer, resuspended in a small volume of buffer, and transferred to an ultracentrifuge bottle. A volume of 1 M KCl was added to give a final concentration of approximately 0.35 M, and the tube was left on ice for 45 min, with periodic mixing. Membranes were removed by a 45-min centrifugation at 100,000g, and the supernatant (nuclear extract) was stored at −70° until used. Cytosols were prepared by centrifugation of the homogenate or the 1000gsupernatant at 100,000g for 45 min.
Ligand Binding Assay.
The ability of each flavone compound to compete with TCDD for binding to the AhR was assessed by incubating aliquots of rat liver cytosol (15 mg protein/ml) or cytosol from untreated Hepa cells (2.1–2.5 mg protein/ml) with a range of seven concentrations (0–1000 nM) of the flavone and [3H]TCDD at 1 nM (rat) or 3 nM (Hepa) for 2 h at room temperature. These concentrations of TCDD are nonsaturating at these protein concentrations. Specific binding of [3H]TCDD was determined in duplicate aliquots by the hydroxylapatite assay (Gasiewicz and Neal, 1982), with correction for nonspecific binding measured in the presence of 150-fold excess of unlabelled 2,3,7,8-tetrachlorodibenzofuran. Data were plotted for each antagonist concentration as a percent of the specific binding of [3H]TCDD in the absence of competitor. IC50 values, representing the concentration of competitor at which specific binding was reduced by 50%, were obtained by nonlinear regression using JMP software (SAS Institute, Inc., Cary, NC).
Electrophoretic Mobility Shift Assay.
Aliquots of rat hepatic cytosol (90 μg protein) or Hepa cell cytosol (21–25 μg protein) from the above incubations with [3H]TCDD, with or without the flavone compounds, were mixed with nonspecific DNA (herring sperm), 0.08 M NaCl, and 25,000–45,000 cpm of [32P]-endlabelled oligonucleotide. The annealed oligonucleotide contained a single consensus DRE that is recognized by the transformed AhR (for complete sequence, see Gasiewicz et al. 1996). Samples were subjected to nondenaturing electrophoresis (4% acrylamide), and [32P] associated with the AhR-retarded band was quantified using a PhosphorImager (PSI, Molecular Dynamics, Sunnyvale, CA). The amount of radioactivity detected at the equivalent position in a lane containing vehicle-treated cytosol was used as background and subtracted from each sample value. Values were expressed as a percent of that observed in cytosol treated with TCDD alone, and IC50 values were determined by nonlinear regression as above.
Cell Transfection and Luciferase Assay.
The antagonist and agonist activities of the flavones were also evaluated in whole cells, using DRE-dependent luciferase as a reporter gene. The reporter plasmid p2Dluc, described previously (Gasiewicz et al. 1996), contained two copies of the DRED consensus sequence (Lusska et al. 1992) and a minimal promoter. LipofectAMINE reagent (Life Technologies) was used to cotransfect 3 μg p2Dluc and 1 μg pGKneo into Hepa cells, according to recommended procedures. Stable transfectants were selected in medium containing 0.9 mg/ml G418 for 10 days. Isolated colonies were expanded and analyzed for luciferase inducibility by TCDD. The subclone designated Hepa-2Dluc.3A4 was used for these studies based on its good inducibility by TCDD and low uninduced levels of luciferase activity. Cells were grown to 80% to 90% confluence in 12-well plates, and triplicate wells were treated with the chosen antagonist (1000 nM), vehicle (DMSO), or 150 pM TCDD, plus a range of antagonist concentrations (0, 1, 5, 10, 50, 100, 500, or 1000 nM). After 4 h incubation, cells were washed twice with PBS, lysed with Reporter Lysis buffer (Promega, Madison, WI), and cells were scraped into microfuge tubes. The final extracts obtained by vortexing and centrifugation of the lysed cells were frozen (−70°) until used to determine luciferase activity using the Promega Luciferase Assay System. Extract was added to luciferase substrate reagent, and light units emitted were immediately measured in a luminometer (Turner Model TD-20e, Turner Designs, Sunnyvale, CA) using a 3-s delay and 15-s integration time. Data were expressed as percent of the light units measured in extracts from TCDD-treated cells, and IC50 values were determined as described above.
Immunoprecipitation and Immunoblotting.
Hepa cell cytosols or nuclear extracts containing the same amount of total protein were adjusted to 0.12 M NaCl or KCl and incubated 2–3 h at 4° with anti-AhR antibody [prepared in rabbits against the N-terminal peptide (Poland et al. 1991) by Multiple Peptide Systems, San Diego, CA]. Complexes were precipitated with Protein A-Sepharose (Pharmacia, Piscataway, NJ), and proteins were separated by SDS-PAGE (6.8% acrylamide resolving gel). Proteins were electrotransferred to Immobilon-P (Millipore, Bedford, MA) using a semidry apparatus (Hoefer Scientific, San Francisco, CA). Samples were loaded onto gels in triplicate sets so that identical pieces of membrane could be probed concurrently with antibodies recognizing Arnt (affinity purified polyclonal, kindly provided by A. Poland), hsp90 (monoclonal from Stressgen, Victoria, BC), and AhR [monoclonal Rpt-1 (Perdew et al. 1995) purified from cell culture supernatant; hybridoma cells kindly provided by G. Perdew]. Membranes were blocked for 1.5 h with 5% Blotto (nonfat dry milk in 50 mM Tris, pH 7.5, 150 mM NaCl, 0.2% v/v Tween 20), incubated with primary antibodies diluted in 1% Blotto for 1.5 h, then for 1 h with appropriate secondary antibodies conjugated to horseradish peroxidase (Jackson Immunoresearch, West Grove, PA). Detection was by chemiluminescence, using reagents purchased from KPL (Gaithersburg, MD) or Amersham (Arlington Heights, IL).
Immunocytofluorescence.
To visualize subcellular localization of AhR, Hepa cells were treated with antagonistII or VIII for 4 h and then exposed to TCDD (1 nM) or DMSO (0.02%) for an additional hour. Cells were fixed and stained for AhR as described (Pollenz, 1996).
Quantitative Structure Activity Relationship (QSAR) Analysis.
Quantitative structure activity relationships for these flavones were analyzed using the 3-dimensional paradigm of comparative molecular field analysis ligand-based modeling (CoMFA) (Tripos, Inc.) similar to that previously described (Cho and Tropsha, 1995; Jones et al. 1996). The geometries of each chemical were fully optimized from a starting structure in which the rings are coplanar and the 3′-methoxy group, if present, is 90° out of the plane. The PM3 Hamiltonian mathematical operator was chosen for optimization because it best reproduces the structures of nitro-containing compounds. The flavone structures were then over-layered based on the best RMS fit of the 1-, 4-, 5 -, and 1′-position atoms of each compound.
Results
Antagonist Activity of Flavones in Rat Liver Cytosol.
The compounds used in this study were designed and synthesized to further test the structure-activity hypotheses generated from our previous analysis of a broader range of structures (Gasiewicz et al. 1996). All of the structures in the current study are flavone derivatives with substituents added primarily at the 3′ and 4′ positions (see Table1 for structures). Initially we tested these compounds in rat hepatic cytosol for their ability to antagonize (1) [3H]TCDD binding to the Ah receptor, and (2) TCDD-induced DRE binding by the AhR. Inhibition of [3H]TCDD binding by a range of concentrations of each compound was measured to determine IC50values (Table 1, Fig. 1). In this table, the compounds have been listed and numbered in order of increasing IC50 for ligand binding. Those compounds containing a 3′-methoxy substitution as well as a 4′ substituent with one or more terminal atoms of high electron density (I–IV) had the highest affinity. Flavones having 4′ substituents that were either less electron-dense (e.g., V,VII, IX, X) or were bulky (XI) had lower affinity. The 3′-methoxy group itself was apparently critical for inhibition of TCDD-elicited DRE binding, i.e., for antagonist activity (compare III and VIII; also others in Gasiewicz et al. 1996; Lu et al. 1996). The 3′-methoxyflavone (4′-unsubstituted) (V) possessed moderate antagonist activity (Table 1); addition of the 4′-nitro or 4′-azido group (II, III) further enhanced the potency. Bulkier substitution at either the 3′ or 4′ position, or groups that are less electron-rich than 4′-NO2 (e.g., 4′-CN or -NH2), were all associated with decreased binding affinity (VI, IX–XI) and a correspondingly decreased ability to inhibit TCDD-induced DRE binding.
Comparative AhR antagonist and agonist activities of flavone derivatives in rat liver cytosol

Dose-response curves of inhibition of [3H]TCDD binding and TCDD-elicited DRE binding by flavones. Representative data for compounds III(3′-OMe-4′-NO2-flavone) and X(3′-OMe-4′-NH2-flavone) in rat liver cytosol are shown. Values are presented as percent of the level observed with 1 nM TCDD alone. In cytosol treated with [3H]TCDD alone, mean values for total binding, nonspecific binding, and specific binding were 1070 dpm, 80 dpm, and 990 dpm, respectively, in 35 μl (0.52 mg protein). Filled symbols are observed ligand binding; open symbols are observed DRE binding; lines are the nonlinear fits to these data (seeMaterials and Methods).
For many of these compounds, the IC50 for DRE binding was close to the IC50 for ligand binding, suggesting that these agents bind to the AhR, but the resulting complex cannot undergo additional transformation, at least under these experimental conditions, to a DRE-binding conformation. CompoundsVIII and IX, up to 1 μM, however, were unable to inhibit TCDD-elicited DRE binding; thus, although these structures blocked [3H]TCDD binding, the resulting complex retained some DRE-binding function. Compound I(3′-methoxy-4′-iodo) was the most potent inhibitor of [3H]TCDD binding (Table 1), and unlikeVIII and IX, low concentrations of Ialso decreased DRE binding in parallel by up to 50% in some experiments. However, at higher concentrations (>100 nM), DRE binding increased toward 100% of TCDD alone (not shown), consistent with the strong agonist-like activity of I. Thus, a meaningful IC50 for DRE binding cannot be calculated.
Stimulation of DRE-Binding by Flavone Compounds in Rat Liver Cytosol.
The ability of each compound to stimulate DRE binding in the absence of TCDD, i.e., function as an AhR agonist, was also determined. The majority of these compounds elicited little DRE binding at 1 μM (Table 1, last column). Compounds I,VIII, and IX, which were ineffective inhibitors of TCDD-elicited DRE binding, were able to partially transform the AhR to a DRE binding complex (20–50% of levels achieved with 3 nM TCDD). Although the 3′-hydroxy substitution decreased AhR affinity ofVIII compared with III, it enabled AhR transformation to a DRE-binding form to a greater extent. The iodo substitution at either the 4′ or 5′ position (I,IX) also conferred greater agonist activity.
Antagonist/Agonist Activity of Flavones in Mouse Hepatoma Cell Cytosol.
We also performed the equivalent evaluations as described above using Hepa 1c1c7 cell cytosol in order to compare the effectiveness of these antagonists in a second species. The Hepa cell cytosol was chosen rather than, for example, C57Bl mouse liver cytosol, because 1), subsequent mechanistic studies were to be perfomed in Hepa cell transfectants in culture, and 2), we found that C57Bl mouse liver AhR is relatively resistant to in vitro transformation to a DRE-binding form (unpublished observations). A subset of the flavone compounds was selected to represent the range of potencies of antagonist and agonist activity that had been observed in the rat.
The rank order of potency of inhibition of [3H]TCDD binding as well as TCDD-elicited DRE binding by the selected compounds was the same in both species (Table2). The three compounds that at 1 μM were able to significantly induce DRE binding by the rat cytosolic AhR (I, VIII, IX) were also effective agonists in Hepa cell cytosol (Table 2). As in rat cytosol, the agonist activity of VIII and IX was consistent with their inability to inhibit TCDD-induced DRE binding even at 1 μM, whileI behaved as described above for rat liver cytosol. The similarity of the rank order of potency of these compounds in rat and mouse cytosol implies that the observed structure-activity relationship may not be species-specific. Obviously there are quantitative differences between the AhRs from these two species but the ligand binding “pockets” appear to be comparable in their acceptance of these chemical structures. Furthermore, the relative effects of the substituents on the flavone parent structure with respect to antagonizing TCDD-elicited DRE binding and permitting partial agonist-like activity is similar between species, at least under in vitro conditions.
Comparative AhR antagonist and agonist activities of selected flavone derivatives in Hepa lclc7 cell cytosol
Effect of Flavone Compounds on TCDD-Induced Transcription in Cultured Cells.
To further assess these compounds as AhR antagonists, we determined their efficacy as inhibitors of TCDD-induced transcription in Hepa cells stably transfected with a DRE-dependent reporter gene, luciferase. Because some substrates including some substituted flavones directly inhibit CYPIA1 activities such as ethoxyresorufin-O-deethylase (Lu et al. 1996; Ciolino et al. 1998), luciferase induction was chosen as a direct measure of AhR-induced transcription. Initial experiments to characterize the dose-responsiveness and time course of luciferase induction by TCDD showed that even the lowest dose tested (0.5 pM) caused approximately 25% increase over solvent control in light units measured after 5 h exposure to TCDD (not shown). Maximal induction (approximately 150-fold) was achieved by 0.5 nM. To evaluate the effectiveness of antagonism, a concentration of 150 pM TCDD was chosen, at which approximately 50% of maximal luciferase induction was achieved. At this concentration, the luciferase activity rose sharply during the first 4 h and remained high during the next 4 h; by 24 h, it was less than half of maximal activity (Fig.2). We have not investigated further the mechanism of this phenomenon, which is in contrast to the sustained induction of CYPIA1 in cells and in vivo (e.g. Gasiewicz et al. 1986).

Time course of induction of luciferase activity in stably transfected Hepa 2Dluc cells by TCDD and its inhibition by 3′-OMe-4′-NO2-flavone (III). Results are means from triplicate wells. Standard deviations are too small to detect for most data points.
We next determined the time course of antagonism for two of the compounds in order to choose an optimal time of exposure for subsequent experiments. TCDD (150 pM), with and without compounds IIIor IX at 1 μM or 0.1 μM, was added to nearly confluent cultures, and triplicate wells were harvested periodically over 24 h to determine luciferase activity. The observed time course of antagonism for both agents was similar: during the first 4 to 5 h of exposure, both concentrations were equally effective and inhibited approximately 95% and 80% (III and IX, respectively) of the TCDD-induced activity (shown for III in Fig. 2). With time, the effectiveness of inhibition diminished: at 0.1 μM, all antagonism was lost by 24 h, whereas at 1 μM, antagonism was maintained to a greater degree (60–70% inhibition compared with TCDD alone at 24 h). One possible interpretation of these observations is that the flavones are inactivated by the cells, presumably through metabolism; at 1 μM, sufficient compound remains even after 24 h to antagonize the TCDD effect, whereas the lower concentration was essentially completely inactivated by 24 h.
Based on these results, we determined an IC50value for each of the selected flavone compounds following the protocol described under Materials and Methods. All of the selected flavones antagonized the ability of TCDD to induce luciferase activity (1st column, Table 3). As in the cell-free systems, II and III had the lowest IC50s together with low or no agonist activity. Whereas a concentration of 1 μM compounds VIII,IX, and XI was unable to inhibit 50% of TCDD-elicited DRE binding in Hepa cytosol (Table 2), all were effective inhibitors of TCDD-elicited induction of luciferase in whole cells. This difference may reflect, in part, accumulation of the lipophilic flavones within the cells to higher concentrations than calculated in the growth medium or in the isolated cytosol. However, there was no overt cytotoxicity from exposure to any of these flavones (Table 3, 3rd column). The substantial agonist activity of I andVIII (as assessed by luciferase induction at 1 μM) correlated with DRE-binding in vitro (compare Table 2, last column with Table 3, 2nd column). The ability of II, III, andIX to apparently elicit some (albeit low level) DRE binding in vitro but not induce luciferase in whole cells, demonstrates that EMSA results are not necessarily accurate predictors of in vivo transcriptional activity and suggests a requirement for more than AhR-DRE binding to activate transcription in a cell or in vivo.
Effect of selected flavone derivatives on luciferase induction in stably transfected Hepa cells
QSAR Analysis.
Partial least-squares analysis was performed using IC50s for inhibition of TCDD-elicited DRE binding versus the steric and electrostatic properties of each compound. The cross-validated q2 value is 0.643 with five components (up to ten components were tested). (Any cross-validated q2 value greater than 0.3 is considered significant (Cramer et al. 1988)). The components (orthonormal vectors) are constructed from the initial complete 3-dimensional representations of the structures, and represent/describe the most important features of the molecules that determine activity. The calculatedr2 value was 0.989, with a probability of r2 being 0 of 0.000; and anF value (n1 = 5, n2 = 9) of 164.7, with a standard error of the estimate of 0.20. Recognizing that this model is based on a limited range of structural variability of the molecules, it predicts with high probability that the 3′-methoxy substituent is important in AhR antagonist activity by contributing a negative charge for electrostatic interaction and steric bulk in a favorable position.
Mechanism of AhR Ligand Binding Inhibition.
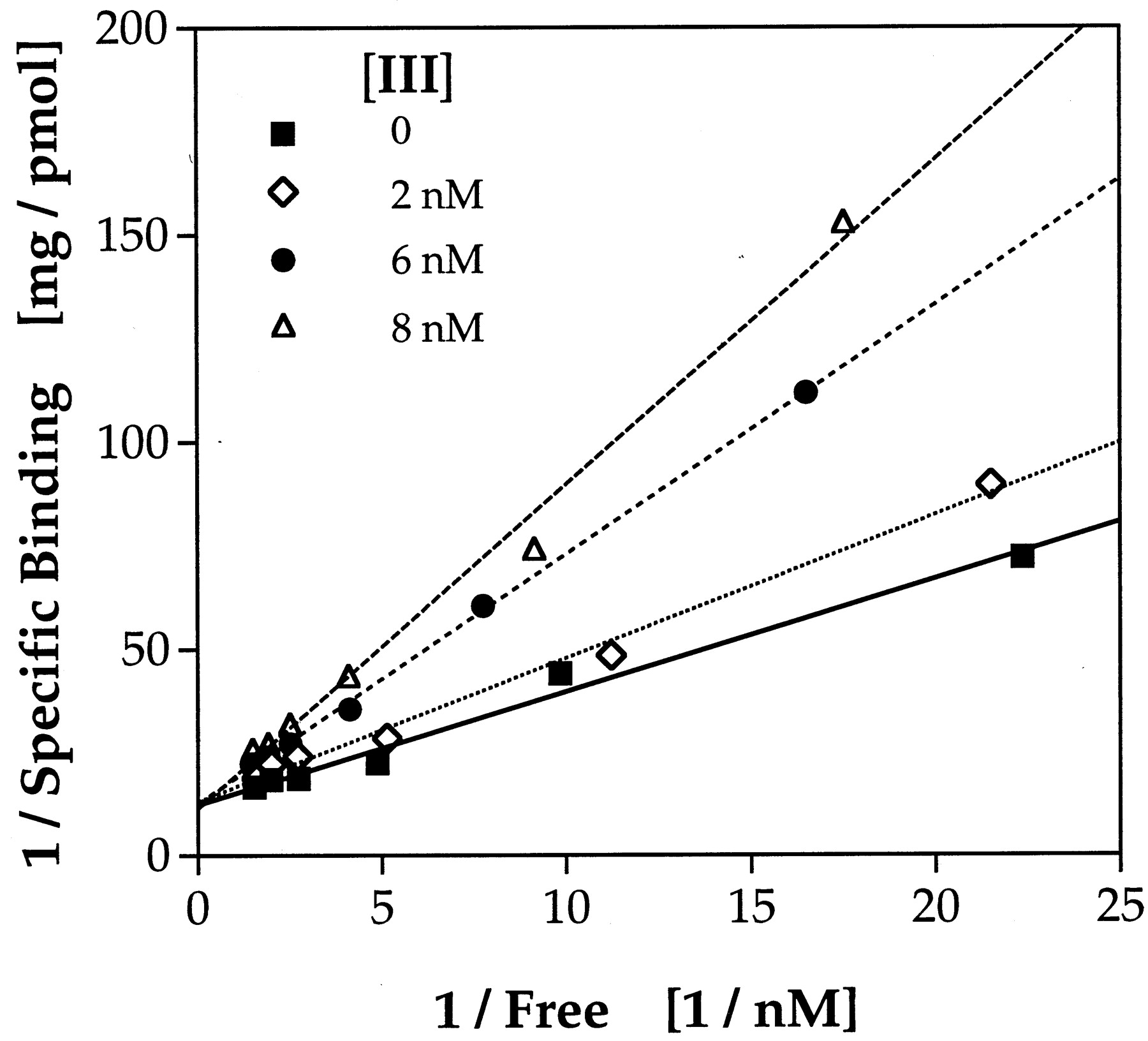
Two of the most potent antagonists were further studied to determine whether their inhibition of TCDD binding to the rat cytosolic AhR was by a competitive or noncompetitive mechanism. Compound II (0, 2, 6, or 8 nM) or III (0, 2, 4, or 6 nM) was added to aliquots of rat liver cytosol (2.5 mg protein/ml) along with a range of concentrations of [3H]TCDD (0.1–1 nM), and specific binding of [3H]TCDD was determined. Double reciprocal plots of the data (1/specifically bound [3H]TCDD versus 1/free [3H]TCDD) gave good fits by linear regression for TCDD alone and in the presence of each concentration of flavone. These data indicate that TCDD and compounds II andIII bind the AhR competitively (shown for III in Fig. 3). Kivalues were calculated from these data: 5.6 ± 1.7 nM forII (compared with the determinedKd for TCDD binding of 0.2 nM for this batch of cytosol); 2.6 ± 0.2 nM for III(Kd for TCDD in this batch of cytosol was 0.4 nM). The observation of competitive binding is strong evidence that these structures bind to the AhR within the TCDD-binding site.

3′-OMe-4′-NO2-flavone is a competitive inhibitor of TCDD binding to rat liver AhR. Rat liver cytosol was incubated with the indicated concentrations of compound IIIand a range of concentrations of [3H]TCDD (0.1–1 nM) for 2 h at room temperature. Specific binding of [3H]TCDD (pmol/mg cytosolic protein) was determined by the hydroxylapatite assay. Linear regression for each line gave r values of 0.992–0.999.
Analysis of Receptor Complex Components in the Presence of Antagonists.
A second approach to determining mechanisms involved in antagonism of TCDD-dependent Ah receptor function was to compare the subunit composition of the receptor complex in the presence of the different ligands. Antagonism could result from a number of possible events following the binding of the flavone in the TCDD-binding site. Some of these are 1), prevention of nuclear uptake of the ligand-bound AhR, 2), prevention of the normal agonist-dependent release of associated proteins such as hsp90 from the complex, 3), prevention of the association of the ligand · AhR with Arnt, 4), formation of a complex that includes Arnt, but that lacks DRE-binding ability, or 5), the ligand · AhR · Arnt complex binds the DRE sequence but lacks transcriptional enhancement activity.
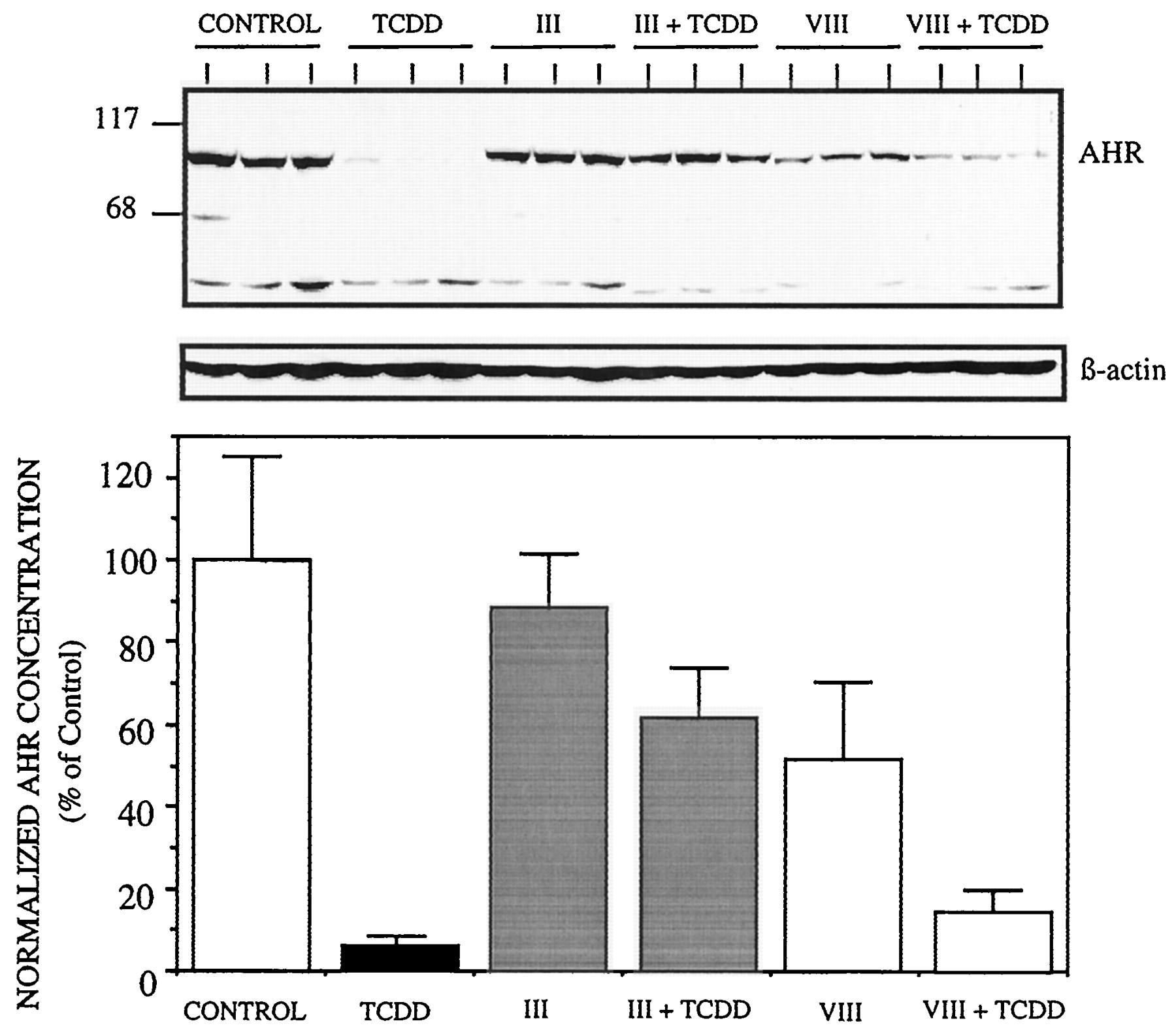
Antibodies to the AhR monomer were used to immunoprecipitate the receptor complex from nuclear extracts or cytosol after treatment of Hepa cells for 1 h with vehicle, TCDD, or the selected flavone derivatives. The precipitated proteins were identified by Western blotting using antibodies against Arnt, hsp90, and AhR, as described inMaterials and Methods. None of these proteins was detected when nonspecific rabbit IgG was used for immunoprecipitation (not shown). When cells were treated with TCDD or B-naphtnoflavone (another AhR agonist), AhR complexes were principally precipitated, as expected, from nuclear extracts (very little from cytosol) and clearly contained Arnt but no hsp90 (Fig. 4, lanes 2, 7). This was in contrast to AhR complexes from untreated cells, which were primarily found in the cytosolic fraction, and contained hsp90 but not Arnt (Fig. 4, lane 1). Flavones II, III, andVIII were chosen for these experiments based on the observation that II and III appeared to be the best antagonists as determined by our various in vitro and whole cell criteria, while VIII was found less effective as an antagonist and possessed some agonist activity. In cells treated withII or III, like vehicle-treated cells, very little AhR and no associated hsp90 were immunoprecipitated from nuclear extracts, although the small amount of receptor that was detected in the nuclear fraction was apparently associated with Arnt (Fig. 4, lanes 3, 5). Most AhR in these cells was immunoprecipitated from cytosolic extracts, in association with hsp90 and not Arnt (Fig. 4, lanes 3, 5). In contrast, after treatment of cells with VIII, more AhR was detected in nuclear extracts, and the coprecipitated Arnt signal in these extracts was strong (lane 6). Thus, VIII seemed to mediate some AhR nuclear translocation and association with Arnt, consistent with its partial agonist activity in vitro and in transfected cells. In coexposure experiments, compound III(lane 4) and compound II (not shown) blocked the ability of TCDD to elicit the dissociation of hsp90 and dimerization with Arnt. The presence of Arnt in immunoprecipitated complexes correlated with strong DRE binding in the nuclear extracts as monitored by EMSA (Fig.4).

Flavone antagonists inhibit hsp90 dissociation from AhR and dimerization with Arnt. AhR complexes in cytosolic and nuclear extracts prepared from Hepa cells exposed for 1 h to vehicle, TCDD, or the indicated flavone compound were immunoprecipitated using an anti-AhR polyclonal antibody. Precipitated proteins were separated by SDS-PAGE and identified by Western blotting. Samples were loaded on the gels in triplicate groups so that identical membranes could be probed with anti-AhR, anti-hsp90, and anti-Arnt antibodies simultaneously. DRE binding was also quantified for these nuclear extracts and is indicated as a percent of the value in extracts from TCDD-treated cells. A representative experiment is shown. For both nuclear (A) and cytosolic (B) samples, Lane 1 = vehicle control (Veh); Lane 2 = TCDD-treated (T); Lanes 3, 5, 6 = compoundIII, II, VIII alone, respectively; Lane 4 = III + TCDD; Lane 7 = BNF; Lane 8 (hsp90 blots only) contained 100 ng purified hsp90.
Effect of Antagonists on AhR Subcellular Localization.
Although the above results are suggestive, they are not proof of altered subcellular AhR localization because proteins redistribute during disruption of cells to obtain nuclear and cytosolic fractions. Therefore, as a more direct method of comparing the effects of agonist and antagonist, we used immunofluorescence microscopy to visualize the AhR in cells treated with III or VIII in the presence and absence of TCDD. Cells were fixed and stained with antibodies specific for the AhR. Consistent with previous studies (Pollenz et al. 1994; Pollenz, 1996), treatment of cells with TCDD resulted in a dramatic redistribution of AhR from the cytosolic to the nuclear compartment (Fig. 5, Panel B compared with A). Exposure to the partial agonist, VIII, also resulted in increased AhR in the nucleus both in the absence and presence of TCDD (Fig 5, Panels E, F). In contrast, exposure of cells to compound III alone did not result in a significant amount of nuclear AhR staining, and the presence of III inhibited the TCDD-induced nuclear translocation (Fig 5, Panels C, D). These results provide strong verification of our tentative conclusions based on the above coimmunoprecipitation results.

Subcellular localization of AhR in antagonist-treated cells. Hepa cells were treated with 1 μM III orVIII for 4 h and then 1 nM TCDD or DMSO (0.02%) for an additional hour. Cells were fixed and stained with A-1 anti-AhR (1 μg/ml) followed by goat anti-rabbit IgG conjugated to Texas Red (1:750). Treatments were as follows: A, DMSO (0.02%); B, TCDD; C, III and DMSO; D, III and TCDD; E,VIII and DMSO; F, VIII and TCDD; G, control cells stained with preimmune IgG (1 μg/ml).
Effect of Antagonists on AhR Protein Degradation.
To further characterize the mechanism of antagonist action on AhR-mediated signaling, the level of AhR proten was determined in Hepa cells exposed to compound III or VIII. Previous studies have shown that TCDD treatment elicits a dramatic down-regulation of AhR protein after entry of the AhR into the nucleus (Pollenz, 1996). Thus, it is hypothesized that a potent antagonist would inhibit this down-regulation. Quantitative Western blot analysis of total cell lysates indicated that AhR protein was substantially depleted following TCDD exposure (Fig. 6). Exposure toVIII alone also resulted in reduced AhR levels that were further reduced after exposure to TCDD. In contrast, exposure of cells to III alone did not significantly reduce AhR levels, and the presence of III significantly inhibited AhR degradation after addition of TCDD (Fig. 6). These observations are consistent with the immunofluorescent staining data and with the finding that nuclear localization of the AhR precedes its down-regulation (Pollenz, 1996).

Antagonist III blocks TCDD-induced down-regulation of AhR protein. Hepa cells were treated with compoundIII or DMSO for 1 h and then TCDD or DMSO for an additional 6 h. Total cell lysates were prepared and resolved by SDS-PAGE. Blots were probed with antibodies to AhR and β-actin. Protein bands were quantified by computer densitometry, and AhR values were divided by those of β-actin. Each bar represents the average and S.D. of three independent plates of cells.
Discussion
The structures synthesized and evaluated for Ah receptor antagonist activity in this study were all based on the flavone ring system as some derivatives of this class were previously found to be potent antagonists (Lu et al. 1995; Gasiewicz et al. 1996; Lu et al. 1996). Of the structures examined in the current report, II(3′-methoxy-4′-azidoflavone) and III(3′-methoxy-4′-nitroflavone) were the most potent antagonists with little agonist activity. Compounds II and IIIboth possess 4′ substitutions that have higher electron density than the other 4′ substituents tested. These data, along with the information obtained from other substituted flavones in this and our previous investigations (Gasiewicz et al. 1996) are consistent with the conclusion that small (to accommodate the 14 × 12 × 5Å ligand-binding pocket) electron-rich centers at the 4′ position promote antagonist activity of the 3′-methoxyflavones. Based on the data presented here, we also hypothesize that the high electron charge density external to the ring structure in the 4′-azido or 4′-nitro compounds (II, III) may permit formation of an external H-bond with the AhR binding site, as illustrated in Fig.7A. Such H-bonding and/or electrostatic interaction between flavone and AhR may stabilize the AhR · ligand complex in a conformation that prevents dissociation of hsp90. In contrast, structures such as VIII–XI, which are poorer antagonists, can form internal H-bonds (Fig. 7B) that would decrease the electrostatic charge at the terminal atoms. Furthermore, the observation that the 3′-methoxy group is critical for potent antagonism is consistent with the possibility that electrons from the methoxy oxygen delocalize by resonance to permit an additional increase in the electron density of the 4′ substituent. It is also possible that electron-rich substitutions at both the 3′ and 4′ positions are necessary for optimal interaction with amino acids of the AhR ligand-binding site. A related structure, 3′-methoxy-2′-aminoflavone (PD98059), was recently reported to inhibit MAP kinase kinase and to antagonize both TCDD binding to the AhR and transformation to a DRE-binding form in rat liver cytosol (Reiners et al. 1998). Reported IC50s for these 3 parameters were 1–4 μM when TCDD was used at 10 nM. However, in preliminary studies using this compound and 1 nM TCDD in rat liver cytosol, we obtained IC50s for ligand binding and DRE binding of 407 nM and 610 nM, respectively, and no agonist-like activity at 1 μM (data not shown). These values are not significantly different from those for X (3′-methoxy-4′-aminoflavone), suggesting that the 3′-methoxy may be the major determinant of antagonism in these low-potency amino-group-containing derivatives.

Illustration of hypothesized external hydrogen bonding between flavones II or III and amino acid residues of the AhR (A) and internal H-bonding in flavonesVIII, IX, and XI (B).
Compound I (3′-methoxy-4′-iodoflavone) had high binding affinity for both rat and Hepa AhR, but was also a relatively potent agonist as defined by induction of DRE binding and luciferase transcription. Several 4′-halogenated flavones (3′-unsubstituted) tested by Lu et al. (1996) also were found to be partial AhR agonists in rat liver cytosol. Unlike the azido or nitro groups, an iodo (or other halogen) substituent cannot form H-bonds with external protons (on the receptor). Thus, as is well-documented for the dioxin and dibenzofuran family, lateral halogenation of some flavone derivatives is also associated with AhR agonist activity.
In general, we observed relatively consistent behavior of the flavones in terms of rank order of potency within our criteria of antagonism/agonism and between species using cell-free systems. However, there was a poorer correlation between inhibition of TCDD-elicited DRE binding in Hepa cell cytosol and inhibition of TCDD-induced luciferase activity in transfected cells. Our experimental protocol for measurement of luciferase induction by TCDD and its inhibition by the flavones was chosen to be as comparable as possible to the in vitro receptor-binding and DRE-binding assays. Nonetheless, intact cells and isolated cytosol obviously provide very different environments for the receptor and these differences likely account for the quantitative disparities in effectiveness of the test compounds between the two systems. For example, (1) the cell membrane provides a barrier between the ligands and the Ah receptor. Differences in lipid solubility among the compounds are likely of little importance in isolated cytosol but would lead to different rates of uptake and retention in the intact cell, and hence differences in their availability to compete with TCDD. (2) In intact cells, Arnt is localized to the nucleus (Pollenz et al. 1994; Holmes and Pollenz, 1997) whereas it is present in cytosolic extracts of cells/tissues untreated with AhR ligand. Thus, in the intact cell, the compartmentalization of the processes of ligand binding and receptor transformation may further magnify small differences among the test compounds in their rates of diffusion, association with AhR, and their abilities to elicit nuclear translocation/transformation. (3) Although metabolic breakdown of the flavones was not specifically investigated, our data (Fig. 2) suggest that it may have a substantial effect on the efficacy of these compounds in intact cells. (4) The test agents were not overtly cytotoxic (Table 3), but they or their metabolites could have some as yet unidentified effect on cellular function/signal transduction pathways etc. unrelated (or related) to their interaction with AhR (e.g., Reiners et al. 1998). This possibility could confound interpretation of the results in whole versus fractionated cells. Despite these factors, the general conclusions from the whole cell and the in vitro results are consistent, namely that II andIII are the most potent antagonists and have low agonist activity.
Based on our competitive binding, coimmunoprecipitation, and receptor localization studies, we propose the following model of action for the potent 3′-methoxyflavone antagonists which have a 4′-nitro or -azido substitution (II, III). These flavones bind in the AhR ligand-binding pocket, and their high binding affinity may reflect an electrostatic interaction and/or H-bonding with receptor amino acids(s). However, whereas TCDD initiates a conformational change in the receptor resulting in nuclear translocation of the liganded AhR as well as loss of hsp90, the 3′-methoxyflavones fail to initiate the conformational change. The flavone · AhR complex primarily remains in the cytosol, associated with hsp90 and perhaps other AhR-associated components such as the immunophilin-like proteins recently identified (Carver and Bradfield, 1997; Ma and Whitlock, 1997 ). The detection of a small amount of AhR in the nucleus (Fig. 5) or AhR · Arnt in nuclear extracts (Fig. 4) after treatment with II orIII suggests that a small proportion of AhR liganded with these compounds may undergo nuclear localization and dimerization with Arnt. However, induction of luciferase transcription was not detected (Table 3). This lack of transcriptional enhancement could be due to an insufficient concentration of the AhR · Arnt complex in the nucleus, and/or the formation of an AhR · Arnt complex that can bind DRE in vitro (Table 2) but is unable to initiate transcription.
We further interpret the data to suggest that this conformational alteration depends on structural features of the ligand in addition to those that confer high affinity binding. Thus, mere binding of a ligand to the AhR is necessary but not sufficient to elicit receptor transformation. In light of our observations, it is interesting that point mutations within the hormone-binding domain of the estrogen and progesterone receptors have differential effects on the binding of agonist compared with antagonist compounds (reviewed by McDonnell et al. 1994). Both the AhR and steroid receptors can accomodate diverse ligand structures; the fact that these structures elicit different receptor functions may reflect their distinct amino acid contact(s) within the ligand-binding site.
In summary, we have evaluated the interaction of a number of substituted flavones with the rat and mouse AhR and have determined that effective antagonism of AhR function depends on the 3′-methoxy group as well as a 4′ substituent with one or more terminal atoms having high electron density, such as a nitro or azido group. The effective antagonist structures bind to the AhR ligand-binding site but fail, possibly because of their formation of a H-bond with the receptor, to elicit the dissociation of hsp90, nuclear translocation of the liganded receptor, and dimerization with Arnt that are mediated by agonists such as TCDD. AhR antagonists, such as those we have identified, as well as similar structures that resist metabolic breakdown, will be useful tools in further defining the ligand-binding “pocket” of the AhR, dissecting the processes involved in AhR-initiated signal transduction, and defining the roles of other AhR-associated proteins.
Footnotes
-
Send reprint requests to: Dr. E.C. Henry, Box EHSC, Department of Environmental Medicine, University of Rochester Medical Center, Rochester, New York 14642. E-mail:henrye{at}envmed.rochester.edu
-
This research was funded by National Institutes of Health Grant ES02515, Center Grant ES01247, and Training Grant ES07026. These data were presented in part at the Society of Toxicology Annual Meeting, March 1998, Seattle, Washington (Toxicol Sci42:383).
- Abbreviations:
- AhR
- aryl hydrocarbon receptor
- Arnt
- Ah receptor nuclear translocator
- DRE
- dioxin responsive element
- EMSA
- electrophoretic mobility shift assay
- hsp90
- 90 kDa heat shock protein
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- SDS-PAGE
- sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Hepa
- hepatoma cells (Hepa lclc7)
- Received September 2, 1998.
- Accepted January 25, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}