


Abstract
The aryl hydrocarbon receptor (AhR) transcription factor is increasingly recognized as functioning in cell cycle control. Several recent reports have shown that AhR activity in the absence of exogenous agonists or presence of the prototypical ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin can affect G1 phase progression in cultured cells. Serum release of serum-starved (G0) 5L rat hepatoma cells triggers transient AhR activation and P4501A1 protein expression concomitant with the G0/G1-to-S phase transition. In contrast, sustained AhR activation in response to TCDD treatment increases p27Kip1 expression in addition to P4501A1, resulting in G1 phase cell cycle arrest. Treating serum-released 5L cells with the alkyne metabolism-based P4501A1 inhibitor 1-(1-propynyl)pyrene results in prolonged AhR activation, enhanced p27Kip1 expression, and G1 phase arrest after serum release. The data are consistent with a cell cycle role for P4501A1 because they show that P4501A1 negatively regulates the duration of AhR action through the metabolic removal of the receptor agonist, thereby preventing AhR-mediated G1 phase arrest.
The eukaryotic PAS domain protein family contains several members that function as sensors of extracellular signals and environmental stresses affecting growth and development (Gu et al., 2000). Among these members, the aryl hydrocarbon receptor (AhR) regulates adaptive and toxic responses to a variety of chemical pollutants, including polycyclic aromatic hydrocarbons and polychlorinated dioxins, most notably 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Historically, studies of the AhR placed an emphasis on understanding the molecular basis for TCDD toxicity. More recently, however, with the advent of AhR knockout mice, evidence that the AhR contributes to normal physiological processes associated with growth and differentiation has mounted (Schmidt et al., 1996; Gonzalez and Fernandez-Salguero, 1998; Lahvis et al., 2000). The AhR is a soluble cytosolic protein in a complex with the chaperone proteins hsp90 and hsp23 and an immunophilin-like protein (Carver and Bradfield, 1997; Ma and Whitlock, 1997; Meyer et al., 1998; Perdew, 1998; Kazlauskas et al., 1999). Upon ligand activation, the AhR translocates into the nucleus, dissociates from the hsp proteins, and binds to DNA response elements (known as DREs or XREs) as a heterodimer with its partner the Arnt protein, which is itself a member of the PAS protein family (Lees and Whitelaw, 1999). The DNA-bound AhR/Arnt dimer recruits cofactors, and the complex modulates expression of target genes (Kumar et al., 1999; Elferink et al., 2001; Beischlag et al., 2002; Wang and Hankinson, 2002).
A number of reports in recent years identified a role for the AhR in cell cycle control, although the precise mechanism remains ill-defined (Ma and Whitlock, 1996; Weiss et al., 1996; Ge and Elferink, 1998; Kolluri et al., 1999; Elizondo et al., 2000; Puga et al., 2000; Tohkin et al., 2000; Elferink et al., 2001). Studies using the mouse hepatoma Hepa 1c1c7 cell line and the AhR-defective variant revealed that the lack of a functional AhR delayed passage through the G1 phase, consistent with a role for the receptor in promoting G1 cell cycle progression (Ma and Whitlock, 1996). Mouse embryonic fibroblasts (MEFs) from AhR-null mice also grow more slowly because ofcell accumulation in the G2/M phase potentially caused by altered expression of the G2/M kinases Cdc2 and Plk (Elizondo et al., 2000). Using AhR-null MEF cells, the AhR was also shown to contribute to p300-mediated induction of DNA synthesis (S-phase progression) by the adenovirus E1A protein involving an as yet undefined mechanism (Tohkin et al., 2000). Collectively, these observations suggest that, in the absence of exogenous ligands, the AhR functions to promote cell cycle progress. In contrast, a concentration of the exogenous agonist TCDD as low as 10 pM inhibited DNA replication and cell proliferation in confluent mouse epithelial cell cultures (Gierthy and Crane, 1984). TCDD also suppressed DNA synthesis in rat primary hepatocytes (Hushka and Greenlee, 1995) and during rat liver regeneration after partial hepatectomy (Bauman et al., 1995). This evidence is consistent with a cell cycle inhibitory role for the AhR. Studies with 5L rat hepatoma cells (AhR-positive) demonstrated that TCDD induces a G1 phase cell cycle arrest not detected in BP8 cells (AhR-negative cells derived from the 5L line after selection for resistance to BaP genotoxicity) (Gottlicher et al., 1990). Ectopic AhR expression in BP8 cells restores the TCDD-induced arrest response (Weiss et al., 1996; Elferink et al., 2001), confirming the receptor's participation in G1 phase cell cycle control. Moreover, the TCDD-induced G1 arrest in 5L cells is attributed largely to increased expression of the cyclin-dependent kinase (CDK) 2 inhibitor p27Kip1 (Kolluri et al., 1999), which binds to and inhibits CDK2 activity necessary for the transition into S phase (Sherr, 1996; Sherr and Roberts, 1999).
A key target for CDK2 activity is the retinoblastoma tumor suppressor protein (pRb) (Weinberg, 1995; Sherr, 1996; Sherr and Roberts, 1999), a major G1 cell cycle checkpoint control protein functionally inactivated by CDK-mediated phosphorylation (Weinberg, 1995). Recent reports demonstrated that pRb interacts with the AhR through two distinct receptor domains (Ge and Elferink, 1998; Puga et al., 2000; Elferink et al., 2001). A cyclin D-like LXCXE motif within the AhR-PAS domain was confirmed by site-directed mutagenesis studies to confer AhR-pRb binding, and this interaction seems to be restricted to the hypophosphorylated “active” form of pRb (Elferink et al., 2001). Functional evidence demonstrated that, in addition to TCDD-induced G1 arrest, maximal CYP1A1 induction in 5L cells relies on pRb binding with the AhR-LXCXE motif (Elferink et al., 2001). Given that the hypophosphorylated pRb is confined to the G0 and G1 phases of the cell cycle, the AhR-pRb interaction—and functional consequences of this interaction—are likely to be cell cycle-dependent.
The seemingly contradictory observation that AhR activity in the absence of exogenous agonists promotes cell growth, whereas TCDD can inhibit growth, provided the impetus for the studies described in this article. The roots for a molecular explanation reconciling this apparent conundrum lie in a recent finding that an endogenous AhR agonist is also a substrate for the P4501A1 enzyme encoded by the CYP1A1 gene (Chang and Puga, 1998). P4501A1-mediated depletion of an endogenous AhR agonist establishes a negative feedback mechanism that suppresses prolonged AhR activity under normal physiological conditions. The evidence presented here suggests that the duration of AhR activity can dramatically impact the cell cycle response to growth factors and other extracellular signals, consistent with the hypothesis that the AhR functions as a modulator of cell cycle progression through G1 phase.
Materials and Methods
Materials. T4 DNA kinase was purchased from Invitrogen (Carlsbad, CA). RNase A was obtained from Sigma-Aldrich (St. Louis, MO). The pRb antibody was purchased from Pierce Biotechnology, Inc. (Rockford, IL); antibodies against mouse AhR and Arnt were kindly provided by Dr. R. Pollenz (University of South Florida, Tampa, FL); the p27Kip1 antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); and the P4501A1 antibody was obtained from BD Gentest (Woburn, MA). The TfR antibody and all horseradish peroxidase-conjugated secondary antibodies were obtained from Zymed Laboratories (South San Francisco, CA). FBS was acquired from Hyclone Laboratories (Logan, UT) or Invitrogen. Dialyzed FBS and charcoal-stripped FBS were purchased from Hyclone Laboratories. TCDD was supplied by the National Cancer Institute Chemical Carcinogen Reference Standard Repository. 1-PP was kindly provided by Dr. W. Alworth (Tulane University, New Orleans, LA). 3′-Methoxy-4′-nitroflavone was kindly provided by Dr. T. Gasciewicz (University of Rochester, Rochester, NY). Radioactive compounds and enhanced chemiluminescence reagents were acquired from Amersham Biosciences Inc. (Piscataway, NJ). Custom synthesized oligonucleotides were obtained from Invitrogen. All other chemicals were purchased from Sigma-Aldrich.
Cell Culture. Wild-type rat hepatoma 5L cells and AhR-defective BP8 variants were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 atmosphere at 37°C. Where indicated, cultures were serum-starved for 24 h in either serum-free media or DMEM containing 0.1% FBS before serum release in DMEM containing 10% FBS.
EROD Activity. EROD activity was determined according to the method described by Kennedy et al. (1993). Subconfluent 5L cultures (100-mm plates) were treated with 10 nM TCDD in DMSO for 24 h before treatment with 1-PP [0.1% (v/v) in DMSO], which was added to the media 15 min before cells were harvested, washed in PBS, and recovered by scraping in 500 μl of 25 mM HEPES, 1.5 mM EDTA, and 10% (v/v) glycerol, pH 7.5. Cells were lysed by freeze-thawing at –80°C, and 50 μl of lysate (1–2 mg/ml of protein) was mixed with 20 μM 7-ethoxyresorufin (25 μl) in 100 mM sodium phosphate, pH 7.8, and incubated at 37°C for 15 min in 96-well plates. Reactions were started by the addition of 25 μl of 4 mM NADPH and stopped after 5 min with 150 μl of acetonitrile (resorufin production was linear with respect to time over this period). Bovine serum albumin was substituted for cell lysates in blank reactions. Fluorescence was read in a Fluoroskan Ascent (Thermo Electron, Waltham, MA) plate reader at 530-nm exitation and 620-nm emission. EROD activity (picomoles of resorufin formed per minute per milligram of protein) was calculated based on the resorufin and protein concentrations determined from standard curves. Each assay was carried out in triplicate, and the results are presented as the mean ± S.D. from at least three independent experiments. Protein concentrations were determined using the bicinchoninic acid method in accordance with the manufacturer's protocol (Bio-Rad, Hercules, CA).
Flow Cytometry. Cells were trypsinized, washed twice in PBS containing 1 g/l glucose and 5 mM EDTA, fixed in ice-cold 70% ethanol at 3 × 106 cells/ml, and stored at 4°C for at least 18 h. Cells were stained with 50 μg/ml propidium iodide and 1 mg/ml RNase A for 30 min in the dark at room temperature. DNA content analyses were performed on a FACSCalibur cytometer using CellQuest and ModFit software (BD Biosciences, Franklin Lakes, NJ).
Western Blots. For total cell lysates, subconfluent cultures (100-mm plates) were washed once in PBS, and the cells were harvested by scraping in 500 μl of SDS-polyacrylamide gel electrophoresis loading buffer and boiled for 10 min. Nuclear extracts were prepared according to the method described by Denison et al. (1988). Protein was fractionated by 8% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane that was blocked for 1 h at RT in 4% (w/v) dry milk in Tris-buffered saline, pH 7.5, and 0.1% (v/v) Tween 20. Membranes were incubated with primary antibodies for 4 h at RT or overnight at 4°C, with horseradish peroxidase-conjugated secondary antibodies for 1 h at RT, and visualized using the enhanced chemiluminescence detection method according to the manufacturer's protocol.
Electrophoretic Mobility Shift Assay. 5L cells were grown to 80 to 90% confluence; after the cytosolic fraction was prepared, EMSA was performed as described in detail by Reiners et al. (1997). Preparation of nuclear extracts and EMSA on these extracts was performed as described by Denison et al. (1989). The complementary oligonucleotides 5′-GATCCGGCTCTTCTCACGCAACTCCGAGCTCA-3′ and 5′-GATCTGAGCTCGGAGTTGCGTGAGAAGAGCCG-3′ contain an AhR-DNA binding site (underlined) that was annealed and end-labeled with [γ-32P]ATP for use as a DNA probe in the assay.
Results
Cycling cells in culture will enter a quiescent (G0) state upon withdrawal of serum from the growth medium. Serum stimulation triggers a synchronized reentry into the G1 phase of the cell cycle, subsequent commitment to DNA replication (S phase), and a new round of cell division (G2/M phase). Using this experimental paradigm, we observed in 5L cells that serum release and passage through G1 phase—as measured by pRb hyperphosphorylation— coincided with a rapid induction of the CYP1A1 gene in the absence of an exogenously added AhR agonist (Fig. 1). Treatment of cells with the high-affinity AhR antagonist 3′-methoxy-4′-nitroflavone (3Me4NF) (Henry et al., 1999) completely suppressed P4501A1 induction consistent with an AhR-dependent response (Fig. 1). Moreover, serum did not induce P4501A1 in the AhR-negative BP8 cells, further implicating the AhR in serum-induced CYP1A1 gene expression. Although serum release activates the AhR in 5L cells, pRb hyperphosphorylation establishes that AhR activity under these conditions fails to trigger the G1 phase arrest detected in 5L cells after TCDD exposure. To address the formal possibility that P4501A1 induction is caused by an AhR agonist contaminating the serum, we assayed for P4501A1 inducibility in charcoal-stripped and dialyzed serum (Fig. 2A). The evidence reveals that the induction response persists with both charcoal-treated and dialyzed serum (10-kDa cut-off) and is indistinguishable from the response with complete serum (Fig. 2A). Moreover, the serum factor responsible for P4501A1 induction is also heat labile (Fig. 2B). Collectively, the data point to the P4501A1 inducer as being ≥10-kDa protein, possibly a serum-derived growth factor(s).
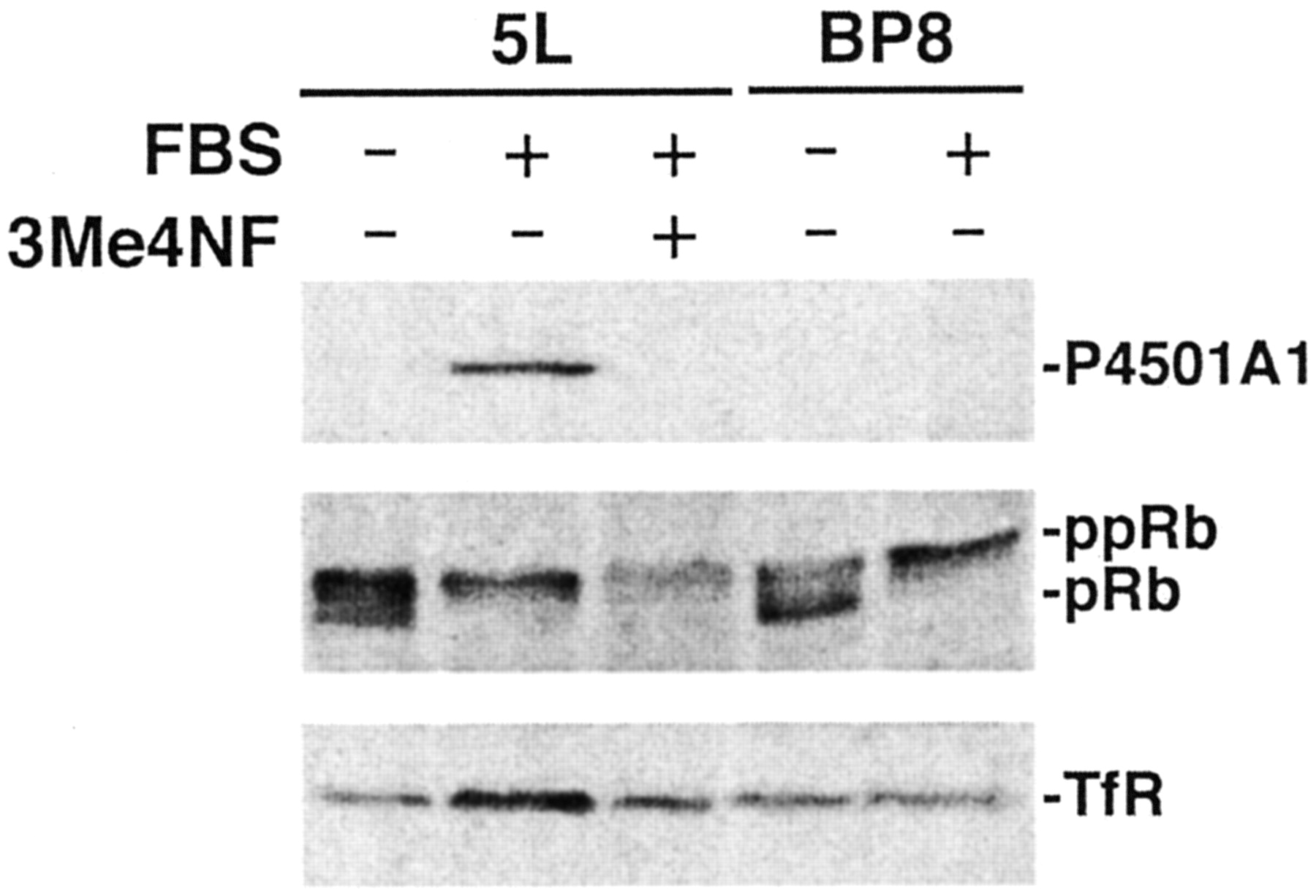
Release of serum-starved 5L cells induces AhR-mediated expression of P4501A1. Subconfluent asynchronous 5L and BP8 cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 0.1% FBS (–FBS) or 10% FBS (+FBS) was added for 4 h in the absence of the AhR antagonist 3Me4NF (–) or presence of 1 μM 3Me4NF (+). Total cell lysates were prepared and analyzed by Western blotting for P4501A1, pRb, and TfR (loading control). Active hypophosphorylated pRb is distinguishable from inactive hyperphosphorylated pRb (ppRb).
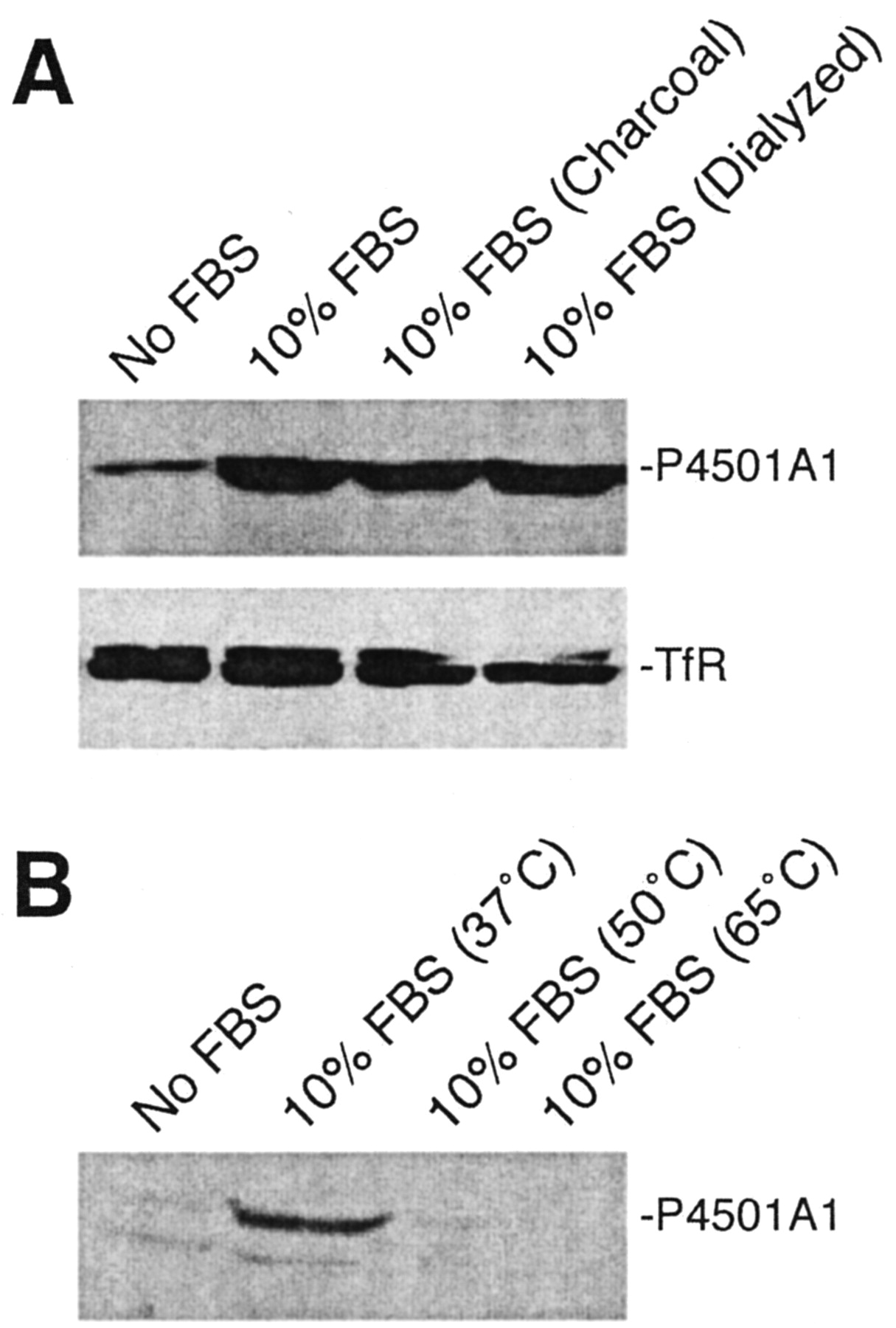
Serum-induced P4501A1 protein expression is growth factor-mediated. A, subconfluent asynchronous 5L cell cultures were serum-arrested in DMEM without FBS for 24 h. Fresh media containing 10% normal FBS, 10% charcoal-stripped FBS, or 10% dialyzed FBS was added for 8 h. Total cell lysates were prepared and analyzed by Western blotting for P4501A1 and TfR. B, serum-arrested 5L cell cultures were released for 8 h with media containing 10% normal FBS treated at the indicated temperature for 1 h, and total lysates analyzed by Western blotting for P4501A1 protein.
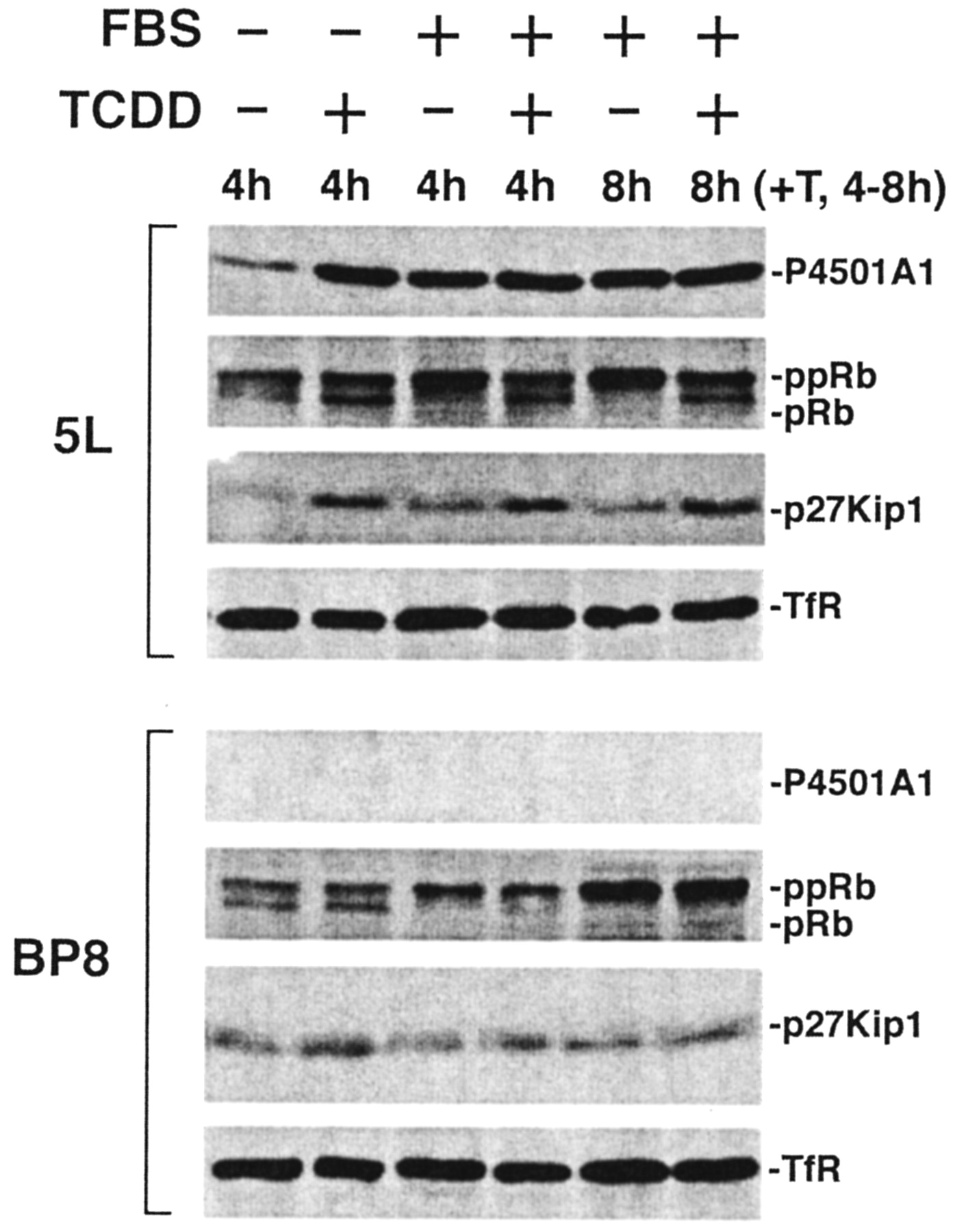
A comparative analysis of the P4501A1 induction response after exposure to 10 nM TCDD or 10% FBS reveals that both stimuli increase P4501A1 expression to a similar degree within 4 h (Fig. 3). In fact, because TCDD and serum in combination fail to increase further CYP1A1 expression, the two stimuli seem to independently trigger a maximal response. However, the two stimuli differ in their effect on pRb phosphorylation and expression of p27Kip1. Hyperphosphorylation of pRb occurs during late G1 phase in preparation for S phase and within 4 h after serum release in 5L cells (Fig. 3, +FBS). In contrast, TCDD treatment prevents pRb phosphorylation (and cell cycle progression) concomitant with increased expression of the CDK inhibitor p27Kip1. Remarkably, TCDD added 4 h after serum stimulation is able to induce p27Kip1 and reverse pRb hyperphosphorylation (Fig. 3, compare lanes 3 and 5 with lane 6). However, in cells released with serum for longer than 5 to 6 h before TCDD exposure, cells become increasingly refractory to the TCDD-induced growth arrest (data not shown), suggesting that TCDD-induced pRb dephosphorylation (and growth arrest) seems confined to late G1 and is lost after commitment to S phase. Hence, although both serum and TCDD can activate the AhR, the nature of the stimulus dramatically influences the growth response. Consistent with previous reports (Kolluri et al., 1999; Elferink et al., 2001), the failure of TCDD to induce p27Kip1 and either prevent or reverse pRb hyperphosphorylation in the AhR-negative BP8 cells supports the conclusion that these processes are AhR-mediated.

AhR activation in response to serum and TCDD induces distinct changes in protein expression. Subconfluent asynchronous 5L and BP8 cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 0.1% FBS (–FBS), 10% FBS (+FBS), or 10 nM TCDD were added for the indicated times. One treatment paradigm (+T, 4–8 h) involved serum release for 4 h followed by TCDD treatment (in the continued presence of serum) for another 4 h. 5L and BP8 total cell lysates were prepared and analyzed by Western blotting for P4501A1, pRb, p27Kip1, and TfR.
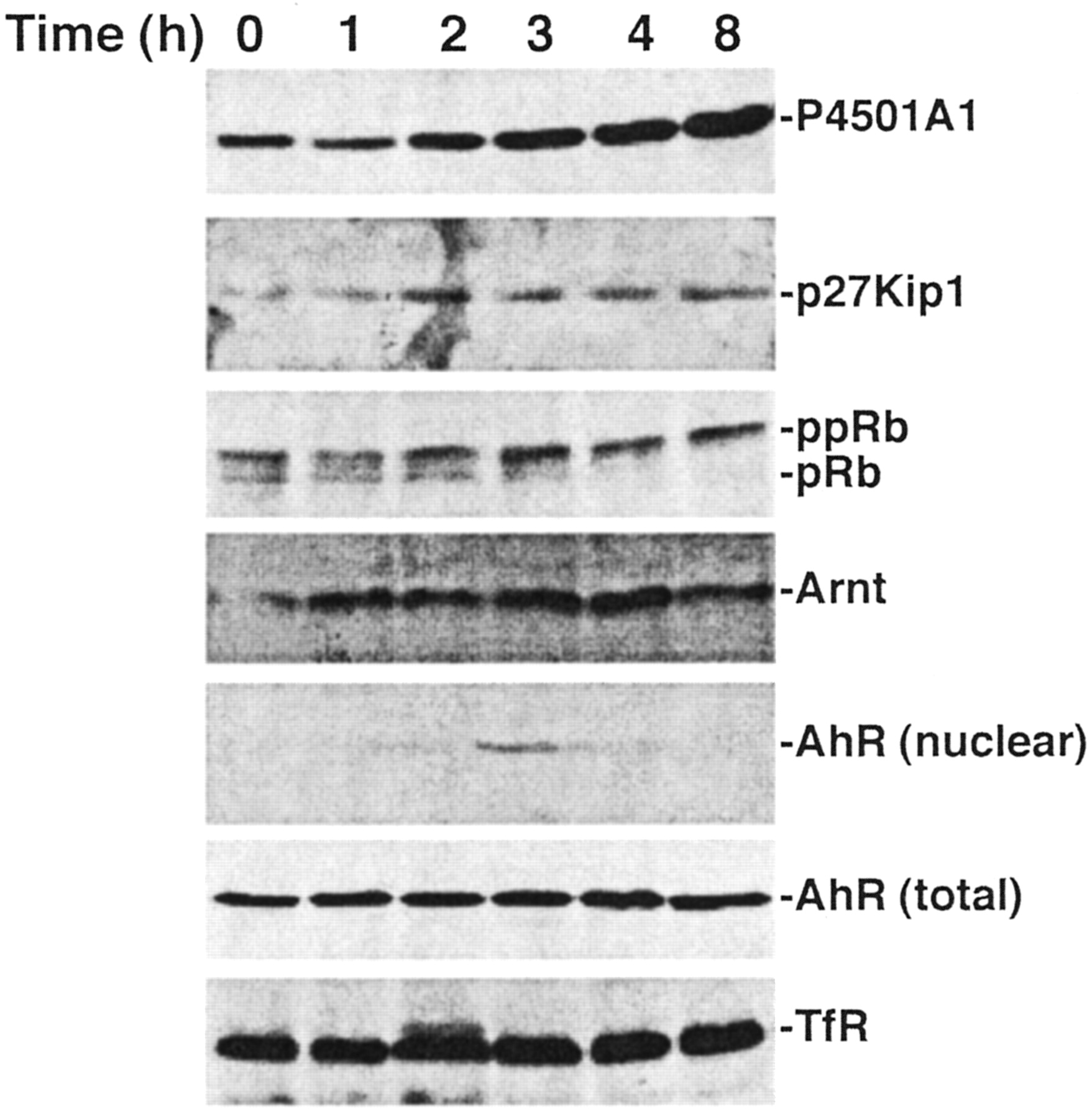
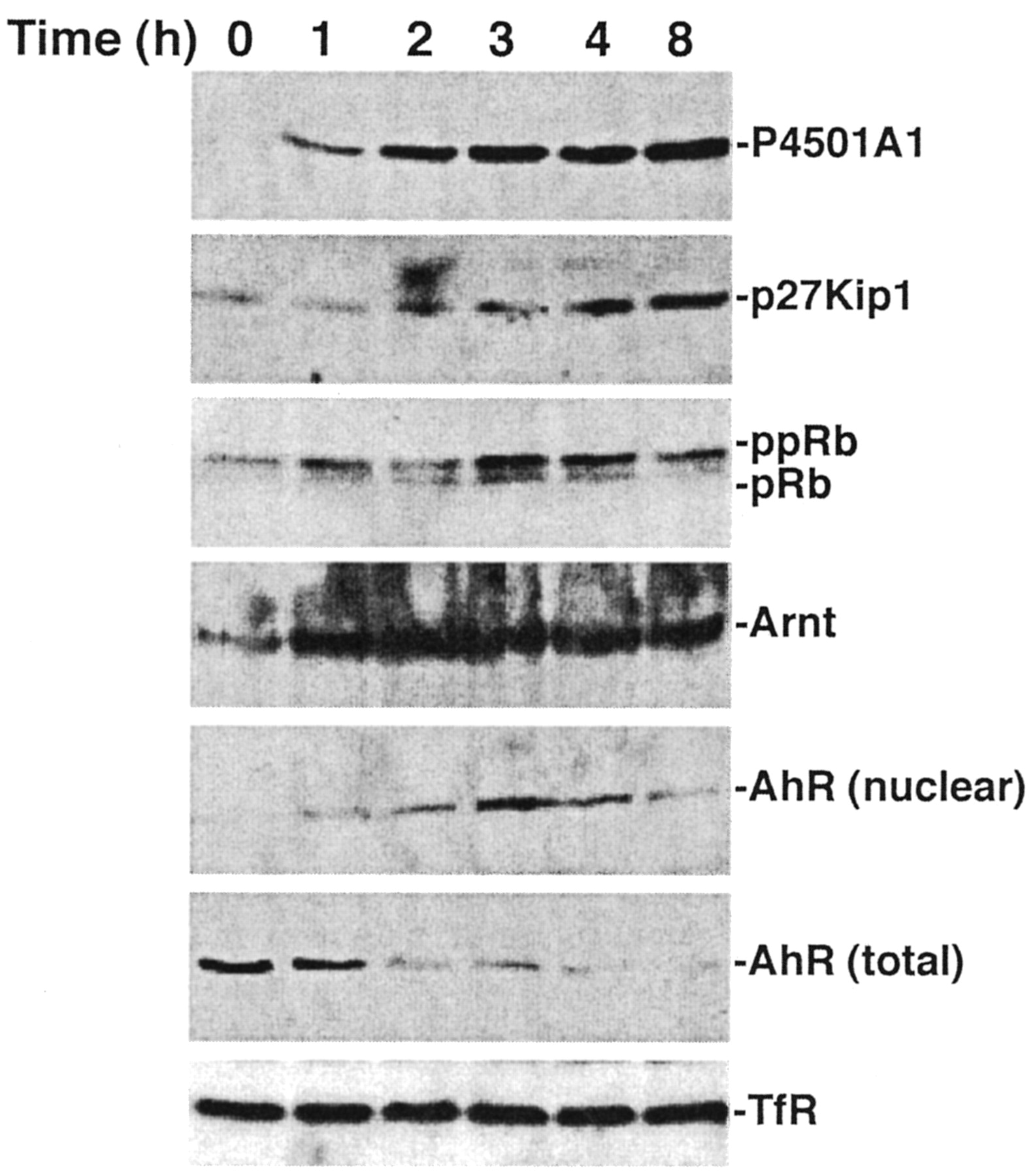
Closer examination of the serum response reveals that the increase in P4501A1 protein is first detectable within 2 h and reaches a plateau between 4 and 8 h (Fig. 4). As measured by nuclear translocation, the P4501A1 increase coincides temporally with AhR activation. Nuclear AhR levels are transient, however, peaking at 3 h and declining rapidly to an undetectable level. Analysis of the Arnt protein in nuclei confirms that the fluctuation in nuclear AhR protein represents entry into and subsequent efflux (or degradation) of the AhR. Detection of the total AhR protein level demonstrates that only a small fraction of the AhR is activated, yet this fraction is able to induce CYP1A1 expression to a degree similar to that of TCDD response (see Fig. 3). Distinct from observations in mouse 3T3 fibroblasts (Vaziri et al., 1996) and PLCH-1 teleost hepatoma cells (Hestermann et al., 2002), AhR expression in 5L cells is not altered by serum withdrawal and restoration (Fig. 4, total AhR). Because only hypophosphorylated pRb binds to the AhR (Puga et al., 2000; Elferink et al., 2001), the loss of nuclear AhR protein coincident with pRb hyperphosphorylation suggests that continued AhR function may depend partly on pRb binding. A similar time course study examining TCDD induction of P4501A1 detects a more rapid increase of P4501A1 protein in keeping with earlier detection of the nuclear AhR protein (Fig. 5). Moreover, nuclear AhR protein remained detectable for the entire 8-h period of the experiment despite the decrease in total AhR protein. The decline in AhR protein presumably reflects ubiquitin-mediated degradation of activated AhR as described previously (Davarinos and Pollenz, 1999). The data also demonstrate that TCDD treatment increases p27Kip1 protein levels and suppresses pRb phosphorylation, presumably through the inhibitory action of p27Kip1 on CDK2 activity (Kolluri et al., 1999). The correlation between p27Kip1 expression and the sustained presence of nuclear AhR protein suggests that induction of p27Kip1 expression and G1 phase cell cycle arrest depends upon prolonged AhR activity.

Serum release triggers transient AhR activation. Subconfluent asynchronous 5L cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 10% FBS were added for the indicated period, and either total cell lysates [P4501A1, p27Kip1, pRb, AhR (total), TfR] or nuclear extracts [Arnt, AhR (nuclear)]were prepared for Western blotting to analyze the indicated proteins. Nuclear extracts were prepared using the method described by Denison et al. (1988).

TCDD induces sustained AhR activation. Subconfluent asynchronous 5L cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 0.1% FBS and 10 nM TCDD were added for the indicated period. Total cell lysates [P4501A1, p27Kip1, pRb, AhR (total), TfR] or nuclear extracts [Arnt, AhR (nuclear)]were prepared for Western blotting to analyze the indicated proteins.
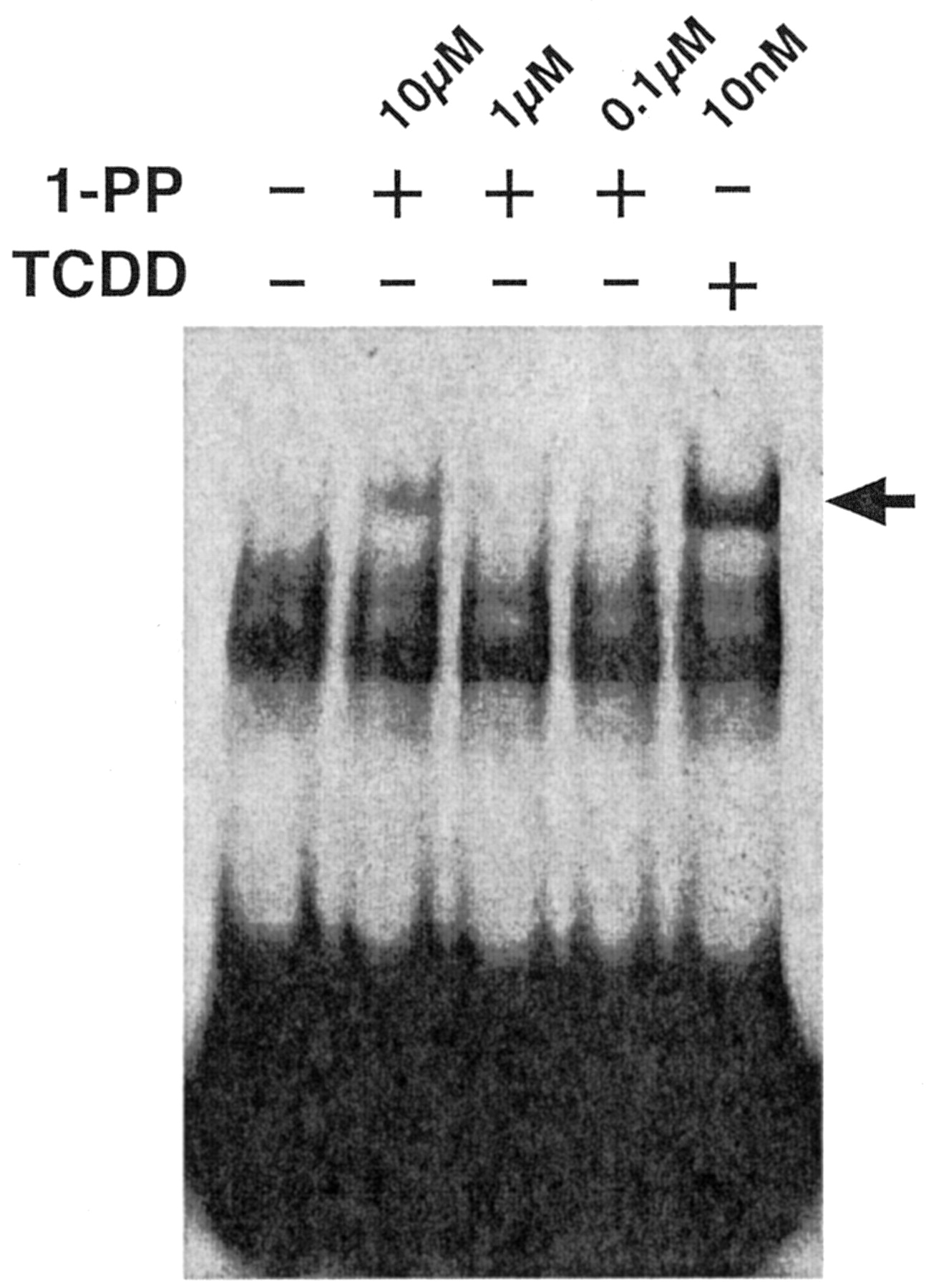
Chang and Puga (1998) proposed that P4501A1 enzyme activity metabolizes an endogenous AhR ligand to an inactive metabolite, creating a regulatory feedback loop that controls AhR activity. This proposal implies that either P4501A1 activity is physiologically important in necessitating tight control over its expression or that P4501A1 activity serves to attenuate transcription of other AhR target genes. In keeping with this latter idea, we hypothesized that serum-mediated induction of CYP1A1 serves to inactivate the AhR, thereby preventing prolonged receptor activity and G1 phase arrest and facilitating commitment to cell division after mitogen stimulation. Hence, a predictable outcome of inhibiting P4501A1 activity would be prolonged AhR activity and G1 phase arrest. To examine this possibility, we experimentally performed cell cycle studies using the P4501A1, 1A2, and 1B1 suicide substrate 1-PP (Shimada et al., 1998). In a recombinant bacterial expression system, the 1-PP IC50 for P450s 1A1, 1A2, and 1B1 are 3 nM, 39 nM, and 90 nM, respectively (Shimada et al., 1998). Because these values were obtained in a reconstituted cell-free system, we first determined the dose response for 1-PP inhibition of EROD activity in intact 5L cells (Fig. 6). Asynchronous 5L cells treated with 10 nM TCDD for 24 h to induce the P4501A1 protein were subsequently treated with 1 nM to 10 μM 1-PP for 15 min before preparing the cell lysates for use in the EROD assay. EROD activity measurements reveal that the IC50 for 1-PP is approximately 150 nM when applied to intact cells in culture and that EROD activity is completely inhibited by 1-PP at concentrations above 1 μM. These values correspond well with 1-PP-mediated P4501A1 inhibition in Hepa1 cells (Alexander et al., 1999). Displacement of [125I]2-iodo-7,8-dibromodibenzo-p-dioxin by 1-PP (EC50 = 22 nM) from the mouse AhR (Alexander et al., 1999) suggests that 1-PP may be a low-affinity receptor agonist. AhR-DNA binding was analyzed by EMSA using the cytosolic receptor from 5L cells transformed in vitro with 0.1 to 10 μM 1-PP or 10 nM TCDD (Fig. 7). The data reveal that treatment with 0.1 and 1 μM 1-PP fails to produce a detectable AhR-DNA complex, whereas 10 μM 1-PP results in modest complex formation, albeit substantially less than TCDD-induced DNA binding. The evidence suggests that 1 μM 1-PP cannot induce AhR activation. It is possible that the formation of receptor-DNA complexes is below the level of EMSA detection yet sufficient to trigger a transcriptional response caused by high intrinsic efficacy (Hestermann et al., 2000). If so, this response would be reminiscent of the robust serum-induced transcriptional response involving only a tiny fraction of the available AhR pool.

Dose response for 1-PP inhibition of P4501A1 activity in 5L cells. Subconfluent asynchronous 5L cell cultures were treated with 10 nM TCDD for 24 h to induce the P4501A1 enzyme. Cultures were subsequently treated with the indicated dose of 1-PP for 15 min before the total cell lysates were prepared as described in Materials and Methods. Lysates were assayed for EROD activity using the method described by Kennedy et al. (1993). The data presented are the mean (± S.D.) of five independent experiments performed in triplicate.
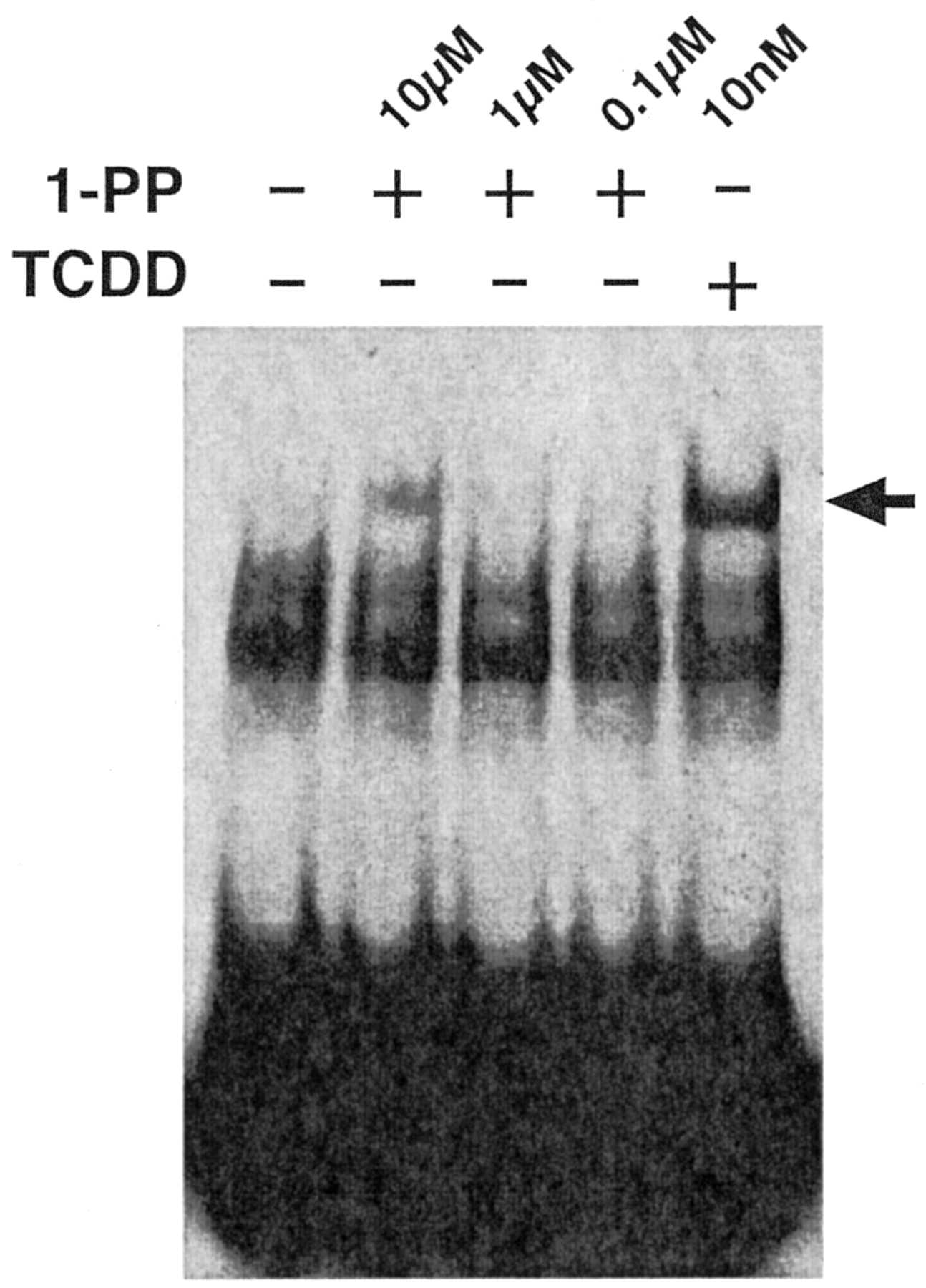
In vitro transformation and DNA binding by the AhR complex in response to TCDD and 1-PP. Cytosol was prepared from subconfluent asynchronous 5L cell cultures as described previously (Elferink et al., 2001) and incubated with DMSO alone (–) or with 0.1 to 10 μM 1-PP or 10 nM TCDD, respectively (+), for 2 h at 20°C. AhR-DNA binding was detected by EMSA as described previously (Elferink et al., 2001). The AhR-DNA complex is denoted by an arrow.
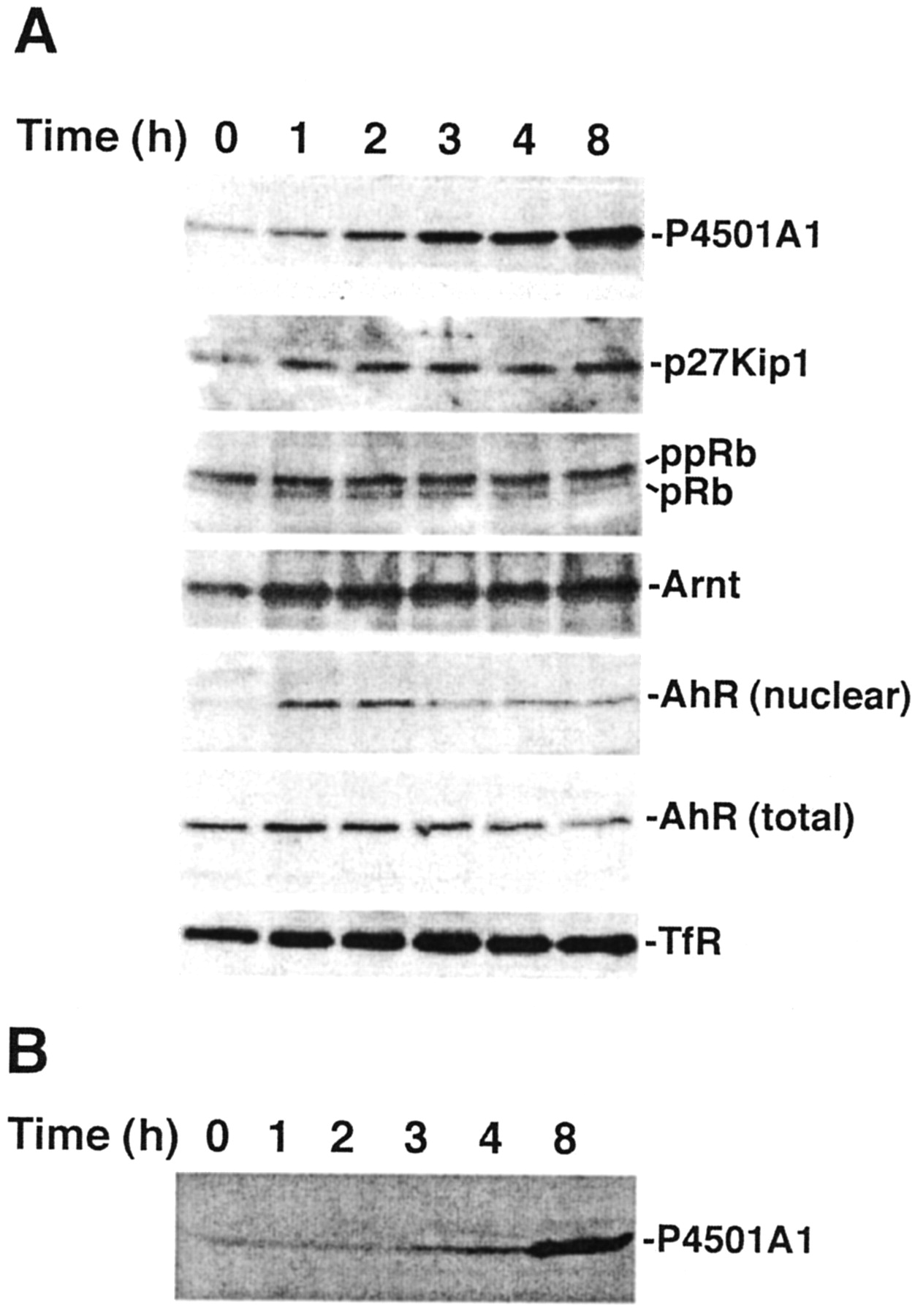
We next tested the effect of 1-PP on CYP1A1 induction and cell cycle progression in serum-stimulated 5L cells (Fig. 8A). Serum-arrested 5L cells were treated with 10% serum in the presence of 1 μM 1-PP for up to 8 h and analyzed by Western blotting. Increases in P4501A1 protein are detectable within 1 h and are akin to the rapid induction response in TCDD-treated cells (Fig. 5) rather than the latent response detected in serum-released cells (Fig. 4). Moreover, the coadministration of 1-PP in serum-released cells results in persistent AhR nuclear localization indicative of sustained AhR activation, again mirroring TCDD response. This prolonged AhR activation also triggered an increase in p27Kip1 protein expression and prevented pRb hyperphosphorylation consistent with a G1 phase cell cycle arrest. Recognizing that 1-PP is a weak AhR ligand, we examined whether 1-PP could induce P4501A1 in the absence of serum stimulation (Fig. 8B). The result indicates that, although P4501A1 is inducible, only a modest increase in P4501A1 protein is detectable after 4 h (which continues to rise by 8 h). In comparison with the TCDD- or serum-mediated CYP1A1 induction response— whether in the presence or absence of 1-PP—the kinetics of CYP1A1 induction by 1-PP alone are inconsistent with direct AhR activation by a low-affinity ligand. This inconsistency strongly suggests that 1-PP is not acting as a classic AhR agonist. Instead, the evidence supports the model that inhibition of P4501A1 activity leads to the accumulation of an endogenous AhR ligand and concomitant AhR activity. The reason for the latent P4501A1 increase is uncertain but may reflect eventual autocrine or paracrine signaling by growth factors released into the culture medium, resulting in a serum-like AhR activation enhanced by 1-PP-mediated P4501A1 enzyme inhibition. 1-PP treatment was neither cytotoxic nor apoptotic to the cells based on trypan blue exclusion and flow cytometric analyses of DNA content, respectively (data not shown).

Serum release in the presence of 1-PP triggers sustained AhR activation. A, subconfluent asynchronous 5L cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 10% FBS and 1 μM 1-PP were added for the indicated period, and either total cell lysates [P4501A1, p27Kip1, pRb, AhR (total), TfR] or nuclear extracts [Arnt, AhR (nuclear)]were prepared for Western blotting to analyze the indicated proteins. B, serum-arrested 5L cells were cultured in fresh DMEM media (without FBS) containing 1 μM 1-PP for the indicated period, and the total cell lysate was analyzed for P4501A1 expression by Western blotting.
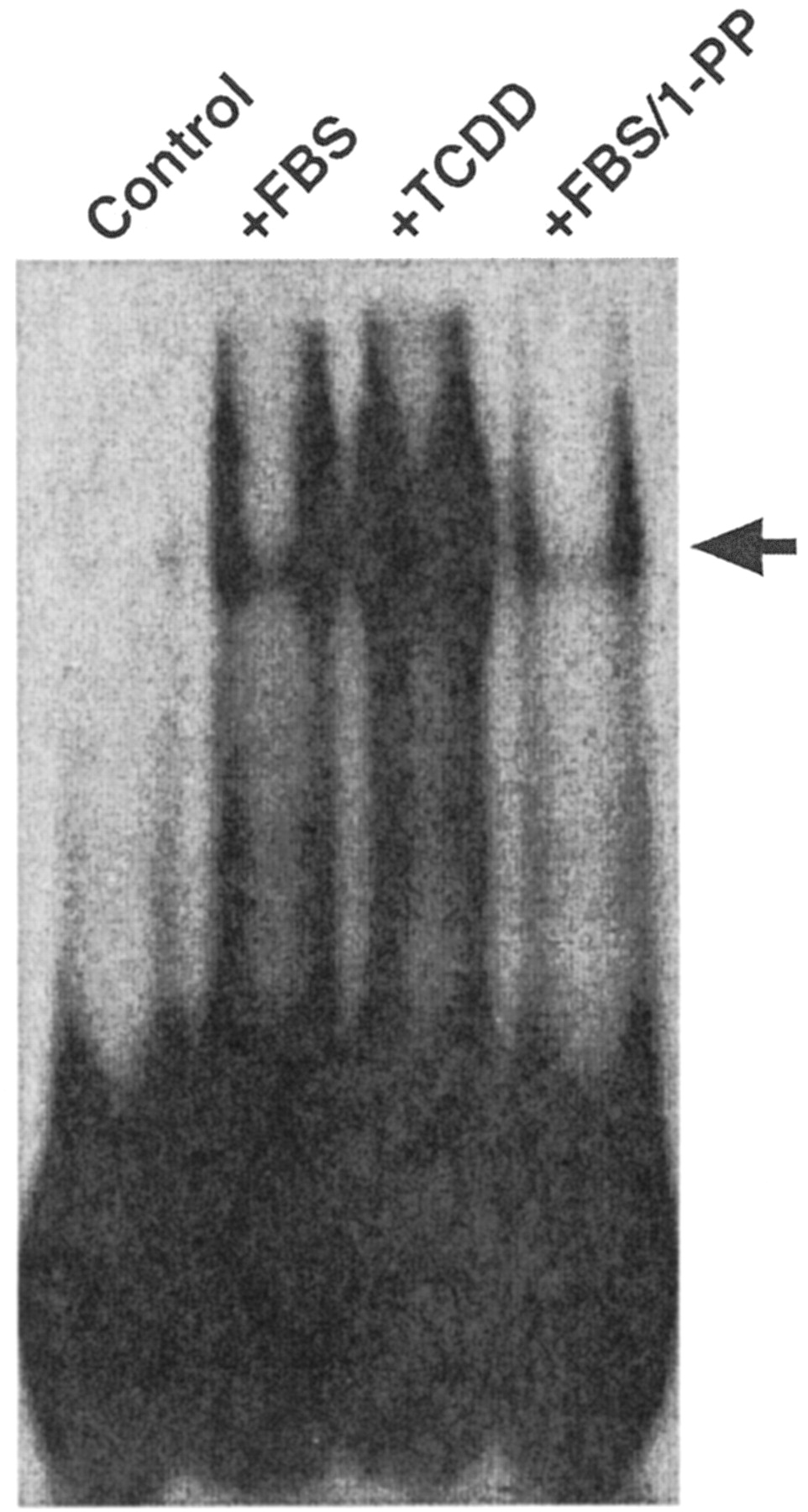
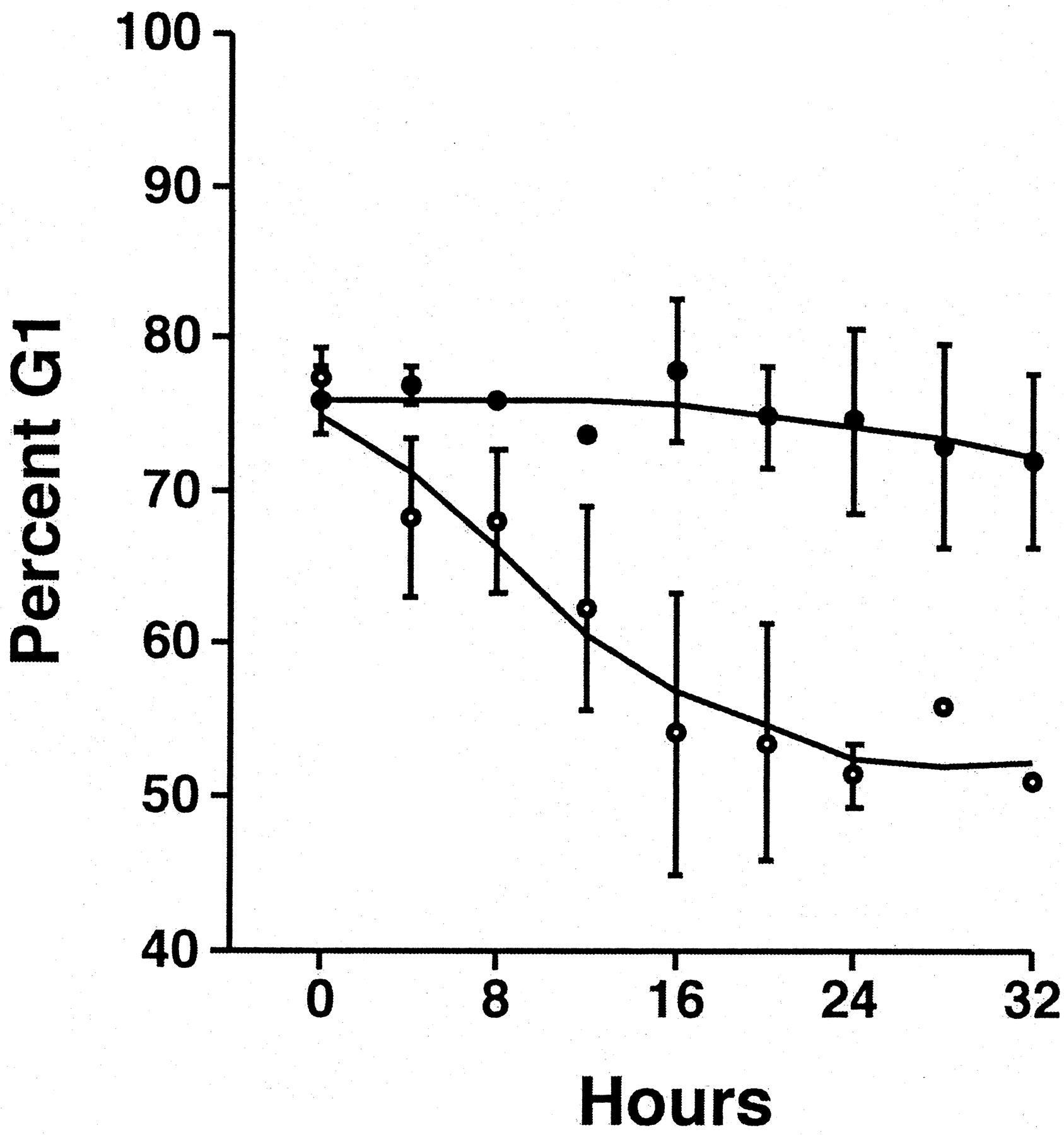
Both the serum-induced increase in P4501A1 protein and AhR nuclear translocation are consistent with a receptor-mediated transcriptional response. However, to confirm that the AhR protein detected in nuclei is functional, we performed EMSA on nuclear extracts from serum-starved cells treated with 10 nM TCDD or 10% FBS in the presence and absence of 1 μM 1-PP for 3 h (Fig. 9). We chose this time point based on the observation that the nuclear AhR level after serum release was maximal at 3 h (Fig. 4). The result confirms that serum treatment induces a readily detectable AhR-DNA complex consistent with receptor-mediated induction of CYP1A1. A further comparison of the serum-mediated induction response in the absence and presence of 1 μM 1-PP over a 20-h period revealed that 1-PP cotreatment progressively increases P4501A1 protein levels above those observed in cells treated with serum alone (Fig. 10). We attribute this increase to prolonged AhR transcriptional activity and the accumulation of 1-PP inactivated P450 protein. Significantly, the effect of 1 μM 1-PP on p27Kip1 expression and pRb phosphorylation in serum-released cells is detectable by 4 h and persists for at least 20 h (Fig. 10). Flow cytometry confirms that 1 μM 1-PP effectively prevents serum-stimulated cells from entering S phase (Fig. 11), consistent with the increase in p27Kip1 protein and pRb phosphorylation status. The data reveal that serum-induced entry into S phase—measured as a pronounced decline in the G1 phase population—is completely absent in serum-stimulated cultures cotreated with 1 μM 1-PP. Additional flow cytometry studies determined that the 1-PP treated cells will eventually exit G1 phase, presumably because of depletion of 1-PP, and that the duration of the arrest response is 1-PP dose-dependent (data not shown).

Serum release induces a DNA-binding AhR complex. Subconfluent asynchronous 5L cell cultures were serum-arrested for 24 h. Cultures maintained in serum-free DMEM (Control), treated with DMEM + 10% FBS/3h (+FBS), 10 nM TCDD (+TCDD), or DMEM + 10% FBS/3 h in the presence of 1 μM 1-PP. Nuclear extracts were prepared and 30 μg of extract was used in the EMSA as described by Denison et al. (1989).

1-PP promotes serum-induced p27Kip1 protein expression and prolongs pRb hypophosphorylation. Subconfluent asynchronous 5L cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Fresh media containing 10% FBS without (–) or with (+) 1 μM 1-PP were added for the indicated period, and total cell lysates were prepared for Western blotting of P4501A1, p27Kip1, pRb, and TfR.

1-PP prevents serum-induced release of arrested cells from G0/G1 phase of the cell cycle. Subconfluent asynchronous 5L cell cultures were serum-arrested (DMEM + 0.1% FBS/24 h). Arrested cells were released using fresh media containing 10% FBS with (•) or without (○)1 μM 1-PP for the indicated period. Cells were trypsinized, washed in PBS, fixed in ethanol, and stained with propidium iodide. DNA content in 2 × 104 cells was determined using a FACSCalibur cytometer equipped with CellQuest and ModFit software. The percentage of cells in G1 is indicated and represents the mean ± S.D. from at least three experiments.
Discussion
Autoregulation of endogenous substrates by metabolizing enzymes is an important mechanism in maintaining homeostasis in biological systems (Nebert 1991). We envision that, in a normal proliferative setting (for instance, during liver mass maintenance through the daily replenishment of lost hepatocytes), AhR activity will contribute to a well orchestrated G1-to-S phase transition. Although CYP1A1 expression is normally low or absent in the largely quiescent (G0) liver, one report demonstrates that, in contrast with several other P450s, the P4501A1 level increases significantly during the regenerative response after partial hepatectomy in rats (Ishizuka et al., 1997). This observation links CYP1A1 expression to a physiologically relevant proliferative response. Based on the evidence presented here, we propose that a critical P4501A1 function is the modulation of AhR activity by regulating the level(s) of a physiological receptor agonist(s). Growth factor-stimulated CYP1A1 induction during the G1-to-S phase transition serves to inactivate the AhR by rapidly depleting the endogenous ligand. Failure to inhibit AhR activity results in elevated p27Kip1 expression and sustained pRb activity culminating in cell cycle arrest. We propose that, in addition to activating mitogenic signaling pathways, growth factor(s) (e.g., in serum) signaling through one or more surface receptors triggers the formation (or release) of a physiological AhR ligand (Fig. 12). AhR activation results in CYP1A1 induction and subsequent removal of the putative agonist. However, the presence of a stable ligand such as TCDD leads to sustained AhR activity that promotes a cell cycle arrest response. Likewise, excessive or prolonged mitogenic signaling resulting in protracted AhR activity may stall G1 phase progression to avoid premature commitment to a new round of cell division. Therefore, in the context of cell proliferation, the AhR seems to function as a G1 phase “throttle control” by balancing P4501A1 activity that promotes cell cycle progression with the inhibitory action of p27Kip1. The requirement for prolonged AhR activity to induce cell cycle arrest may reside with differential target gene responsiveness. The exquisite transcriptional responsiveness of the CYP1A1 gene after transient AhR activation is attributed to the potent enhancer region comprising multiple XREs (Whitlock, 1993). In contrast, analysis of 1.6 kilobases of the p27Kip1 promoter sequence reveals only one candidate XRE (Kolluri et al., 1999), suggesting that the p27Kip1 promoter is much less responsive to the AhR, necessitating the prolonged AhR activity to increase p27Kip1 expression.

Model depicting the proposed mechanism whereby the AhR regulates transition through G1 phase of the cell cycle. Growth factor(s) signal transduction through cell surface receptors results in formation of a labile endogenous AhR agonist by an undetermined mechanism. P4501A1-mediated depletion of the endogenous agonist establishes a negative feedback loop preventing prolonged AhR activity, thus promoting G1 phase cell cycle progression. Exposure to a stable exogenous agonist (e.g., TCDD) or a condition that stabilizes a normally labile agonist will lead to sustained AhR signaling and expression of p27Kip1, culminating in a G1 phase cell cycle arrest.
The observation that serum can induce endogenous CYP1A1 gene expression has also been reported in CaCo2 (Guigal et al., 2000) and HepG2 cells (Guigal et al., 2001), suggesting that this induction is a general response. However, using transient transfections, Guigal and coworkers (2000, 2001) detected 3-methylcholanthrene-inducible but not FBS-inducible expression from a CYP1A1 promoter-driven chloramphenicol acetyltransferase reporter construct, prompting these researchers to conclude that serum-induced CYP1A1 expression is AhR-independent. This conclusion contradicts our data correlating serum responsiveness with both AhR expression and function (Figs. 1 and 3). Given that both pRb and BRG-1 are AhR binding proteins that promote CYP1A1 induction and associate with chromatin remodeling factors (Brehm et al., 1998; Ge and Elferink, 1998; Puga et al., 2000; Elferink et al., 2001; Wang and Hankinson, 2002), we speculate that chromatin structure contributes to AhR-mediated gene expression. Because transfected reporter constructs do not faithfully reconstitute DNA packaging of an endogenous gene, transcriptional responses dependent on chromatin structure may not be accurately captured by ectopic reporter constructs.
As illustrated in Fig. 12, we attribute serum-mediated activation of the AhR to an extracellular growth factor(s). The evidence indicates that the inducing agent is larger than 10 kDa, heat labile, and resists removal with charcoal (Fig. 2), properties reflective of a protein rather than a small organic molecule such as indirubin (Adachi et al., 2001). It seems unlikely that an extracellular growth factor is a direct AhR agonist, suggesting instead that growth factor signaling at the cell surface activates production of an AhR ligand. In this context, the delay in serum-induced nuclear AhR accumulation (Fig. 4) supports the existence of intermediate signaling steps and contrasts the rapid TCDD-induced AhR activation (Fig. 5). By inhibiting both basal and induced P4501A1 activity with 1-PP, serum-stimulated AhR activation is both more rapid and prolonged (Fig. 8A). The evidence against receptor activation by 1-PP comes from the failure to detect AhR-DNA binding induced by 1 μM 1-PP (Fig. 7) and the absence of rapid CYP1A1 induction typical of canonical AhR agonists (Fig. 8B). Identification of the growth factor(s) or characterization of the physiological AhR agonist will provide important clues into the signaling events responsible for serum-induced AhR activation.
The studies with 1-PP strongly suggest that P4501A1 participates directly in promoting cell growth in a mechanism distinct from its normally perceived role in activating procarcinogens (Shimizu et al., 2000). Yet Cyp1a1–/– knockout mice are viable and lack any obvious phenotype (Dalton et al., 2000). The normal phenotype suggests that either the CYP1A1 gene is not developmentally important for cell growth in vivo or supports the existence of functional redundancy possibly involving P4501A2 or -1B1 activity. The 1 μM 1-PP concentration used in the experiments was reported to inhibit P4501A2 and 1B1 (Shimada et al., 1998). However, P4501B1 is not normally expressed in hepatic cells (Alexander et al., 1999), and the cross-reacting anti-P4501A1 antibody used did not detect the P4501A2 isozyme, suggesting that the 1-PP-mediated cell cycle effect in 5L cells was limited to P4501A1 inactivation. Given that the AhR regulates expression of these P450s, phenotypic defects observed in AhR–/– knockout mice may reflect defects in the expression of multiple cytochromes P450 with overlapping specificities for the endogenous ligand(s). This could include the CYP2S1 gene, a newly identified AhR target gene (Rivera et al., 2002). It will be interesting to determine whether mice defective for multiple cytochromes P450 exhibit growth abnormalities.
Investigations with the synthetic antitumor agent DF 203 determined that its antitumor activity depended on P4501A1 activity (Chua et al., 2000). Labeling studies revealed that metabolism of DF 203 by P4501A1 generates a reactive-intermediate capable of covalently binding to and inactivating the enzyme (Chua et al., 2000). Therefore, the antitumor property of DF 203 may actually be caused by P4501A1 inactivation akin to the action of 1-PP resulting in sustained AhR activity. The evidence suggests that DF 203 and 1-PP seem to curtail cell growth through a common mechanism (i.e., targeting P4501A1 activity). The broad substrate specificity displayed by P4501A1 is consistent with the action of these structurally diverse compounds and may benefit the design of clinically useful antitumor drugs.
Acknowledgments
We thank Drs. W. Alworth and T. Gasciewicz for providing the 1-PP and 3Me4NF reagents, respectively, and Dr. R. Pollenz for the AhR and Arnt protein antibodies. We also thank Dr. K. Mitchell for discussion during manuscript preparation.
Footnotes
-
C.J.E. was supported by National Institutes of Environmental Health Sciences (NIEHS) grant R01-ES07800 and in part by NIEHS Center Grant ES06676.
-
ABBREVIATIONS: PAS, Per/Arnt/Sim; AhR, aryl hydrocarbon receptor; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; DRE, dioxin response element; XRE, xenobiotic response element; Arnt, aryl hydrocarbon receptor nuclear translocator; MEF, mouse embryonic fibroblast; CDK, cyclin-dependent kinase; pRb, retinoblastoma protein; FBS, fetal bovine serum; 1-PP, 1-(1-propynyl)pyrene; DMEM, Dulbecco's modified Eagle's medium; EROD, ethoxyresorufin O-dealkylase; DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; RT, room temperature; EMSA, electrophoretic mobility shift assay; 3Me4NF, 3′-methoxy-4′-nitroflavone; DF 203, 2-(4-amino-3-methylphenyl) benzothiazole; TfR, transferrin receptor.
-
↵1 Present address: Karmanos Cancer Institute, Wayne State University, Detroit, Michigan.
- Received July 7, 2003.
- Accepted October 22, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}