


Abstract
Transitional cell carcinoma (TCC) of the bladder comprises 2% of diagnosed canine cancers. TCC tumors are generally inoperable and unresponsive to traditional chemotherapy, indicating a need for more effective therapies. BRAF, a kinase in the mitogen-activated protein kinase (MAPK) pathway, is mutated in 70% of canine TCCs. In this study, we use BRAF mutant and wild-type TCC cell lines to characterize the role of BRAF mutations in TCC pathogenesis and assess the efficacy of inhibition of the MAPK pathway alone and in combination with other gene targets as a treatment for canine TCC. Analysis of MAPK target gene expression and assessment of extracellular signal-regulated kinase (ERK) 1/2 phosphorylation following serum starvation indicated constitutive MAPK activity in all TCC cell lines. BRAF mutant TCC cell lines were insensitive to the BRAF inhibitor vemurafenib, with IC50 values greater than 5 μM, but exhibited greater sensitivity to a paradox-breaking BRAF inhibitor (IC50: 0.2–1 μM). All TCC cell lines had IC50 values less than 7 nM to the mitogen-activated protein kinase kinase (MEK) 1/2 inhibitor trametinib independent of their BRAF mutation status. ERK1/2 phosphorylation decreased after 6-hour treatments with MAPK inhibitors, but rebounded by 24 hours, suggesting the presence of resistance mechanisms. Microarray analysis identified elevated expression of the ErbB family of receptors and ligands in TCC cell lines. The pan-ErbB inhibitor sapitinib synergized with BRAF inhibition in BRAF mutant Bliley TCC cells and synergized with MEK1/2 inhibition in Bliley and BRAF wild-type Kinsey cells. These findings suggest the potential for combined MAPK and ErbB receptor inhibition as a therapy for canine TCC.
Significance Statement The results of this study (1) identify a novel combination strategy for canine bladder cancer treatment: targeting the ErbB/MAPK signaling cascade and (2) establish the utility of canine bladder cancer as a naturally-occurring model for human MAPK-driven cancers.
Introduction
Transitional cell carcinoma (TCC) is the most common bladder cancer in humans and dogs, comprising approximately 4% and 2% of diagnosed malignancies in each species, respectively (Knapp and McMillan, 2013; Siegel et al., 2018). Most canine TCCs are muscle-invasive tumors of intermediate to high grade, with metastases present in 15% of patients at diagnosis and 50% at death. Canine TCC tumors are typically located in the trigone of the bladder, preventing complete surgical resection in most cases (Knapp and McMillan, 2013). These tumors are treated with cyclooxygenase inhibitors alone or in combination with cytotoxic chemotherapeutics; however, median survival time is typically less than a year for all treatment options, indicating a need for more effective therapies (Knapp and McMillan, 2013; Fulkerson and Knapp, 2015). Canine and human TCCs share similarities in their molecular markers, sites of metastasis, and response to chemotherapeutic agents (Fulkerson et al., 2017). Muscle-invasive bladder cancer is less common in humans than dogs but has a similar poor prognosis (Knowles and Hurst, 2015).
A major distinction between canine and human TCC is the occurrence of activating BRAF mutations in 70% of canine tumors (Duval et al., 2014; Decker et al., 2015; Mochizuki et al., 2015). BRAF is a serine/threonine protein kinase in the mitogen-activated protein kinase (MAPK) signaling pathway that regulates cell growth, proliferation, differentiation, and apoptosis (Dhillon et al., 2007). Mutations resulting in dysregulation of the MAPK pathway occur in one-third of human cancers, with activating BRAF mutations identified in 50% of malignant melanomas and at lower frequencies in colorectal and thyroid carcinomas (Dhillon et al., 2007; Dankner et al., 2018). Ninety percent of activating BRAF mutations in human cancers are valine-to-glutamic acid missense mutations at amino acid 600 (V600E) in the protein’s activation loop (Dhillon et al., 2007). This alteration allows BRAF to signal as a monomer independent of upstream RAS activation, resulting in increased MAPK pathway activity (Dankner et al., 2018).
Several small-molecule inhibitors have been developed to target the MAPK signaling cascade. Vemurafenib, an ATP-competitive inhibitor of BRAFV600E, showed promising antitumor activity in humans with late-stage melanoma, with a 48% response rate compared with 5% with the standard-of-care dacarbazine (Chapman et al., 2011). Despite vemurafenib’s initial success, the majority of these tumors eventually acquired resistance (Sosman et al., 2012). Combination therapies that include both BRAF inhibitors and inhibitors of mitogen-activated protein kinase kinase (MEK) 1/2, BRAF’s downstream target, have shown greater success in melanoma than single-agent treatment, leading to Food and Drug Administration approval of these combinations for the treatment of metastatic melanoma (Ascierto et al., 2016; Long et al., 2017). Unlike melanoma, colorectal tumors are innately resistant to BRAF inhibition (Kopetz et al., 2015). Various resistance mechanisms to BRAF inhibition have been identified in human melanoma and colorectal cancer including upregulation of receptor tyrosine kinases (RTKs), secondary mutations in RAS, and increased signaling through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B pathway (Nazarian et al., 2010; Mao et al., 2013).
The discovery of a homologous BRAF-activating mutation in canine TCC identifies a compelling new potential drug target for canine TCC treatment; however, additional in vitro evaluation of canine BRAF’s function and sensitivity to targeted agents is required. The utility of canine cancers as a model for human cancers has become increasingly widespread. Mouse models are poor predictors of anticancer drug efficacy in human patients. Spontaneous tumors in dogs develop under normal immunosurveillance, share molecular and histologic features with human cancers, and undergo the processes of metastasis and drug resistance. Additionally, clinical trials in dogs provide the opportunity to evaluate the efficacy of novel treatments in chemotherapy-naive patients in a shorter amount of time compared with clinical trials in human patients (Gordon et al., 2009).
In this study, we further characterize BRAF mutations in canine TCC cell lines. We assess the ability of BRAF and MEK1/2 targeted agents to inhibit TCC cell growth and block extracellular signal-regulated kinase (ERK) 1/2 phosphorylation as a measure of MAPK pathway activation. We use differential gene expression analysis to determine other potential gene targets for TCC treatment, identifying combined inhibition of the MAPK pathway and the ErbB family of receptors as a therapy with synergistic activity in both BRAF mutant and wild-type TCC cell lines. The results of this study not only identify a novel therapy for canine TCC, but also establish canine TCC’s value as a model for human MAPK-driven cancers, where clinical trials in dogs with naturally occurring bladder tumors can inform therapies for human patients.
Materials and Methods
Cell Lines.
Human cell lines were provided by Dr. John Tentler, University of Colorado Anschutz Medical Campus (RKO, HT29, and Colo205) or purchased from ATCC (A375) (Manassas, VA). Canine TCC cell lines were provided by Dr. Steve Dow at Colorado State University (Bliley) or Dr. Elizabeth McNiel at Tufts University (Angus1, Kinsey, Tyler1, and Tyler2). All cell lines were maintained in Dulbecco’s modified Eagle’s media supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 mM sodium pyruvate (Thermo Fisher Scientific, Waltham, MA). Cells were incubated at 37°C in 5% CO2 and 100% humidity. Canine cell lines were validated using short tandem repeat analysis with the Canine Stockmarks Genotyping Kit (Thermo Fisher Scientific) as previously described (Supplemental Table 1) (O’Donoghue et al., 2011).
Sequencing of Canine BRAF.
Total RNA was extracted from the Bliley TCC cell line using the RNeasy Mini Kit (Qiagen, Germantown, MD) and reverse transcribed to cDNA with the ImProm-II Reverse Transcription System (Promega, Madison, WI). Polymerase chain reaction (PCR) amplification of BRAF was performed using the following primers: forward, 5′-CACCATGGAAGCCCTATTGGACAAGTTTGGT-3′; reverse, 5′-CTTGAAGGCTGCAAATTCTCCGTA-3′. The resulting amplicon was gel extracted with the QIAquick Gel Extraction Kit (Qiagen) and cloned into an expression vector using the pcDNA/3.2/GW/D-TOPO Expression Kit (Thermo Fisher Scientific). Following transformation into One Shot TOP10 Chemically Competent Escherichia coli (Thermo Fisher Scientific), individual clones were isolated and sequenced at the Proteomics and Metabolics Facility at Colorado State University.
Next, 5′ rapid amplification of cDNA ends (5′ RACE) with the SMARTer RACE 5′/3′ Kit (Takara Bio, Kusatsu, Japan) was performed according to the manufacturer’s protocol. First-strand cDNA was synthesized from total RNA isolated from the Bliley TCC cell line, and 5′ RACE PCR using the reverse primer 5′-GATTACGCCAAGCTTTGGCGTGTAAGTAATCCATGCCCTGTGC-3′ and SeqAmp DNA Polymerase (Takara Bio) was performed to obtain a product containing the 5′ sequence of BRAF. The 5′ RACE PCR product was gel extracted using the NucleoSpin Gel and PCR Clean-Up Kit (Takara Bio) and cloned into the 5′ RACE vector with the In-Fusion HD Cloning Kit (Takara Bio). The resulting construct was transformed into Stellar Competent Cells (Takara Bio) according to the manufacturer’s protocol and individual clones were isolated for sequencing by GENEWIZ (South Plainfield, NJ).
Cell Viability Assays.
Vemurafenib and 5-(2-cyclopropylpyrimidin-5-yl)-3-[3-[[ethyl(methyl)sulfamoyl]amino]-2,6-difluoro-benzoyl]-1H-pyrrolo[2,3-b]pyridine (PLX7904) are inhibitors of BRAFV600E. Selumetinib and trametinib are inhibitors of MEK1/2. Sapitinib is an inhibitor of the ErbB receptors epidermal growth factor receptor (EGFR), ERBB2, and ERBB3. All inhibitors were purchased from Selleck Chemicals (Houston, TX) and stock solutions were prepared in DMSO and stored according to the manufacturer’s protocol.
Cell lines were plated in 96-well plates at 1000–5000 cells/well in 100 μl of complete media and incubated for 24 hours at 37°C. Serial dilutions of inhibitor or DMSO control were prepared in complete media at a 2X concentration and 100 μl was added to each well. Plates were incubated at 37°C for 72 hours and cell proliferation was monitored using the IncuCyte ZOOM Live-Cell Analysis System (Essen BioScience Inc., Ann Arbor, MI). The percentage of confluence at 72 hours was normalized by dividing by confluence at 0 hours, and the relative cell number was determined as a fraction of the DMSO control. Dose-response curves were fitted in GraphPad Prism (version 7) using nonlinear regression of the logarithm of inhibitor concentration versus fraction of control. The IC50 values were determined as the concentration of inhibitor corresponding to one-half the fraction of control on the dose-response curves.
For combination treatments, relative cell number was determined similar to single-agent therapies, and fraction affected was determined as a fraction of DMSO control. Combination indices were calculated for each combination using the CalcuSyn (version 2.11) software from BIOSOFT. Representative images of combination treatments were obtained using red-labeled cells (NucLight Rapid Red Reagent; Essen BioScience Inc.). For both single-agent and combination drug sensitivity assays, three to five independent experiments were conducted for each cell line. Each independent experiment included three technical replicates for each drug concentration and DMSO control.
Western Blotting and Antibodies.
Cells were lysed by sonication in radioimmunoprecipitation assay buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP40, 1% sodium deoxycholate, and 50 mM NaF) supplemented with protease and phosphatase inhibitors (2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin). Lysates were centrifuged at 14,000 rpm to isolate protein fractions and total protein was quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of total protein were resolved on 4%–20% Criterion TGX Protein Gels (Bio-Rad Laboratories, Hercules, CA) and transferred to polyvinylidene difluoride membranes with the TransBlot Turbo Transfer System (Bio-Rad Laboratories). Membranes were blocked in Tris-buffered saline with 0.1% Tween-20 and 5% bovine serum albumin for 1 hour at room temperature. Blots were incubated overnight at 4°C with primary antibodies diluted in blocking buffer [phospho-p44/42 MAPK (Thr202/Tyr204) Rabbit mAb #4370 (1:1000), p44/42 MAPK Rabbit mAb #4695 (1:1,000), and B-Raf Rabbit mAb #14815 (1:1000) obtained from Cell Signaling Technology, Danvers, MA; and α-tubulin #T5168 (1:5000) obtained from Sigma-Aldrich, St. Louis, MO]. Membranes were washed three times with Tris-buffered saline with 0.1% Tween-20, incubated with secondary antibody [goat anti-rabbit IgG horseradish peroxidase conjugated (1:10,000) or goat anti-mouse IgG horseradish peroxidase conjugated (1:10,000) obtained from Bio-Rad Laboratories] at room temperature for an hour, followed by three washes with Tris-buffered saline with 0.1% Tween-20. Blots were developed using Clarity Western ECL Blotting Substrate (Bio-Rad Laboratories) and imaged with a Chemi Doc XES+ System (Bio-Rad Laboratories).
Microarray Analysis.
Total RNA was isolated from cell lines with the RNeasy Mini Kit (Qiagen) and microarray analysis was performed at the Functional Genomics Facility at University of Colorado Denver Anschutz Medical Campus using GeneChip Canine Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA) (Fowles et al., 2017). Gene expression values were determined following robust multiarray average normalization in R (version 3.3) using the oligo package (Halper-Stromberg et al., 2011). Differentially expressed genes were determined using a Benjamani-Hochberg false discovery rate cutoff of 0.05 and a fold change of at least 1.5. If multiple probe sets for a single gene were present, the probe set with the highest variance across samples was used.
MAPK Pathway Activity Score.
MAPK pathway activity scores (MPASs) were calculated as previously described based on expression levels of 10 MAPK target genes: CCND1, DUSP4, DUSP6, EPHA2, EPHA4, ETV4, ETV5, PHLDA1, SPRY2, and SPRY4 (Wagle et al., 2018). Briefly, z-scores for MPAS genes were determined across all samples using log2-transformed expression values. MPASs for each sample were calculated as MPAS = (sum of z-scores for MPAS genes)/(√10).
Results
Canine BRAF Is Homologous to Human BRAF.
Reverse transcription PCR was used to amplify BRAF’s coding sequence from the Bliley TCC cell line. The resulting amplicon, corresponding to predicted canine BRAF AA10-772 (ENSCAFP00000005841), exhibited 99% homology to human BRAF AA62-767 (ENSP00000419060).
Since the predicted canine protein was truncated at the amino-terminus compared with human BRAF, 5′ RACE was used to determine the N-terminal sequence of canine BRAF, revealing an additional 48 amino acids not present in the predicted canine BRAF sequence (ENSCAFP00000005841) (Supplemental Fig. 1). The resulting full-length predicted BRAF protein is 763 amino acids and exhibits 98% homology to human BRAF (ENSP00000419060) (Supplemental Fig. 2). Our analysis also identified a heterozygous V-to-E missense mutation at amino acid 596, consistent with previously identified BRAF mutations in TCC tumors (Duval et al., 2014; Decker et al., 2015; Mochizuki et al., 2015). Western blot analysis of BRAF in canine TCC cell lines and the human BRAF V600E A375 melanoma cell line suggests that BRAF is expressed at a similar size and abundance in both humans and dogs (Supplemental Fig. 3).
Canine TCC Cell Lines Exhibit Constitutive MAPK Activity.
BRAFV600E mutations result in constitutive MAPK activity in human cancers (Dhillon et al., 2007). A gene expression signature quantifying relative MAPK activity in a variety of human cancers was recently described (Wagle et al., 2018). The MPAS was calculated based on expression levels of 10 gene targets of the MAPK pathway obtained using Canine 1.0ST arrays. MPASs were calculated for five TCC cell lines as well as 30 other canine cancer cell lines in the Flint Animal Cancer Center cell line panel (Fowles et al., 2017). TCC cell lines exhibited high MPAS values relative to other canine cancer cell lines, suggesting high MAPK pathway activity in TCC cell lines (Fig. 1, A and B). To validate these findings, MPASs were also calculated using gene expression levels from Canine 2.0 arrays (Fowles et al., 2017). MPASs correlated between Canine 1.0ST and Canine 2.0 arrays (R = 0.9263, P < 0.0001, Fig. 1C).

Canine TCC cell lines exhibit constitutive MAPK pathway activity. (A) Heat map of z-transformed expression values for MPAS genes in the FACC cell line panel. Asterisks (*) indicate TCC cell lines. (B) MPAS values for FACC cell lines. TCC cell lines are shown in red. (C) MPAS values determined using Canine 1.0ST arrays vs. those determined using Canine 2.0 arrays (Pearson r = 0.9263, P < 0.0001). (D) Serum starvation of canine TCC cell lines followed by western blot analysis of ERK1/2 phosphorylation. BRAF mutant cell lines are shown in blue and wild-type are shown in red.
To determine whether canine TCC cell lines with BRAF mutations exhibit constitutive MAPK pathway activity, cells were cultured for 24 hours in the absence of FBS followed by assessment of ERK1/2 phosphorylation by western blot. Five TCC cell lines were analyzed: three with heterozygous BRAF V596E mutations (Bliley, Tyler1, and Tyler2), one with a heterozygous KRAS G12D mutation (Angus1), and one wild-type for BRAF and KRAS (Kinsey) (Das S, Idate R, Cronise KE, Gustafson DL, and Duval DL, unpublished data). ERK1/2 phosphorylation was sustained under serum-starved conditions in BRAF and KRAS mutant cell lines (Fig. 1D). Interestingly, the Kinsey cell line, which does not harbor any known activating cancer gene mutations in the MAPK pathway, also showed constitutive ERK1/2 phosphorylation.
BRAF Mutant Canine TCC Cell Lines Are Insensitive to Vemurafenib, but Sensitive to a Paradox-Breaking BRAF Inhibitor.
The effect of BRAF inhibition on TCC cell proliferation was determined using vemurafenib, an ATP-competitive inhibitor of mutant BRAF. Drug sensitivity assays were also conducted in human BRAF mutant melanoma (A375) and colorectal cancer cell lines (RKO, HT29, and Colo205) with varying degrees of sensitivity to vemurafenib (Yang et al., 2012). All TCC cell lines had IC50 values greater than 5 μM (Fig. 2A; Table 1). Canine TCC cell lines were roughly 10- to 100-fold less sensitive than human BRAF mutant A375, Colo-205, and HT29 cancer cell lines and exhibited levels of sensitivity similar to the RKO colorectal cancer cell line (Table 1).

BRAF mutant TCC cell lines are sensitive to paradox-breaking PLX7904, but insensitive to vemurafenib. Cell lines were treated with serial dilutions of (A) vemurafenib or (C) PLX7904 for 72 hours. Relative viability at each dose was determined as a fraction of vehicle control. Three to five independent experiments with three technical replicates were conducted for each cell line. Error bars represent the S.D. of the fraction of control from combined experiments (n = 3–5). TCC cell lines were treated with 15 μM vemurafenib (B) or 2 μM PLX7904 (D) for 6 (top) and 24 (bottom) hours and assessed for ERK1/2 phosphorylation via western blot analysis. Western blot analysis of Kinsey cell lysate in (B) was performed on a separate blot. BRAF mutant and wild-type canine cell lines are shown in blue and red, respectively. BRAF mutant human cell lines are shown in green.
IC50 values for MAPK inhibitors in canine and human cell lines
To determine whether vemurafenib inhibits MAPK pathway activity in TCC cell lines, ERK1/2 phosphorylation was assessed following 6- and 24-hour treatments with vemurafenib. ERK1/2 phosphorylation was suppressed at 6 hours in BRAF mutant cell lines, but rebounded by 24 hours (Fig. 2B; Supplemental Fig. 4). Similarly, colorectal cancer cell lines achieve MAPK pathway reactivation by 24 hours post-treatment, whereas melanoma cell lines maintain pathway suppression (Corcoran et al., 2012). Conversely, vemurafenib treatment increased ERK1/2 phosphorylation in KRAS mutant Angus1 and BRAF/KRAS wild-type Kinsey cells. This response is consistent with paradoxical activation of the MAPK pathway following BRAF inhibition in KRAS mutant and BRAF/KRAS wild-type human cancer cell lines. Mutant BRAF signals as a monomer, but wild-type BRAF requires dimerization with other RAF isoforms. Binding of BRAF inhibitors to wild-type BRAF stabilizes the formation of dimers, resulting in increased MAPK signaling (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010).
Since BRAF mutations in canine TCC cell lines are heterozygous, we wanted to investigate whether TCC insensitivity to vemurafenib could be due to paradoxical MAPK pathway activation as a result of the wild-type copy of BRAF. Thus, sensitivity to the paradox-breaking BRAF inhibitor PLX7904 was determined in canine and human cell lines. BRAF mutant canine cell lines had IC50 values ranging from 0.2 to 1 μM, similar to BRAF mutant human cell lines, whereas wild-type cell lines exhibited IC50 values greater than 5 μM (Fig. 2C; Table 1). ERK1/2 phosphorylation decreased in BRAF mutant cell lines following 6-hour incubation with PLX7904, but rebounded by 24 hours, although the extent of rebound was less than that with vemurafenib (Fig. 2D; Supplemental Fig. 4). ERK1/2 phosphorylation remained unchanged in KRAS mutant Angus1 and increased in BRAF/KRAS wild-type Kinsey cells.
Canine TCC Cell Lines Are Sensitive to MEK Inhibition.
To determine whether MAPK inhibition downstream from BRAF is an effective therapeutic option, we targeted MEK in canine and human cell lines with the selective, allosteric MEK1/2 inhibitors selumetinib and trametinib. Canine TCC cell lines exhibited similar degrees of sensitivity to MEK inhibition as human BRAF mutant cell lines. Canine TCC IC50 values ranged from 18 to 390 and 0.1 to 6.2 nM for selumetinib and trametinib, respectively. Human cell lines exhibited IC50 values of 81 to 1700 and 0.3 to 8.6 nM for selumetinib and trametinib, respectively (Fig. 3, A and C; Table 1). BRAF mutant cell lines in the Genomics of Drug Sensitivity in Cancer database have median IC50 values of 640 and 19 nM to selumetinib and trametinib, respectively (Yang et al., 2013). Thus, canine TCC cell lines exhibited sensitivities to MEK inhibition similar to human BRAF mutant cell lines. Interestingly, BRAF/KRAS wild-type Kinsey cells were the most sensitive to MEK inhibition of all tested cell lines. This response is supported by Kinsey’s MPAS value, which is the fourth highest in the Flint Animal Cancer Center cell line panel. ERK1/2 phosphorylation was blocked in all cell lines after 6 hours of MEK inhibition; however, as with BRAF inhibition it showed a degree of rebound by 24 hours, indicating reactivation of the MAPK pathway despite MAPK inhibition (Fig. 3, B and D; Supplemental Fig. 4). Combined BRAF and MEK inhibition synergized in BRAF mutant Bliley cells, but not wild-type Kinsey cells (median combination index of 0.5 vs. 1.1, respectively) (Supplemental Fig. 5).

Canine TCC cell lines are sensitive to MEK1/2 inhibition. Cell lines were treated with serial dilutions of selumetinib (A) or trametinib (C) for 72 hours. Relative viability at each dose was determined as a fraction of vehicle control. Three to five independent experiments with three technical replicates were conducted for each cell line. Error bars represent the S.D. of the fraction of control from combined experiments (n = 3–5). TCC cell lines were treated with 500 nM selumetinib (B) or 25 nM trametinib (D) for 6 (top) and 24 (bottom) hours and assessed for ERK1/2 phosphorylation via western blot analysis. BRAF mutant and wild-type canine cell lines are shown in blue and red, respectively. BRAF mutant human cell lines are shown in green.
ErbB Signaling Is Upregulated in TCC Cell Lines Relative to Other Canine Cancer Cell Lines.
Synthetic lethality has been widely explored as an antitumor strategy. Cancer cells often harbor specific oncogenic alterations that may not be targeted effectively alone, but when targeted in combination with a second gene elicit a lethal response (O’Neil et al., 2017). Single-agent treatment of canine TCC cell lines with BRAF or MEK inhibitors yielded an initial attenuation of MAPK pathway activity followed by a rebound in pathway activity by 24 hours of treatment. This short-lived response suggests that MAPK inhibition may not be effective as a monotherapy for TCC treatment. Thus, we sought to identify a second gene target that, when inhibited in combination with BRAF, exhibits a synergistic response. To identify potential candidate targets, we determined genes differentially expressed in TCC cell lines relative to other canine cancer cell lines. To avoid histotype-specific genes, we limited our analysis to 719 cancer-related genes present in the COSMIC database (version 83) (Forbes et al., 2017).
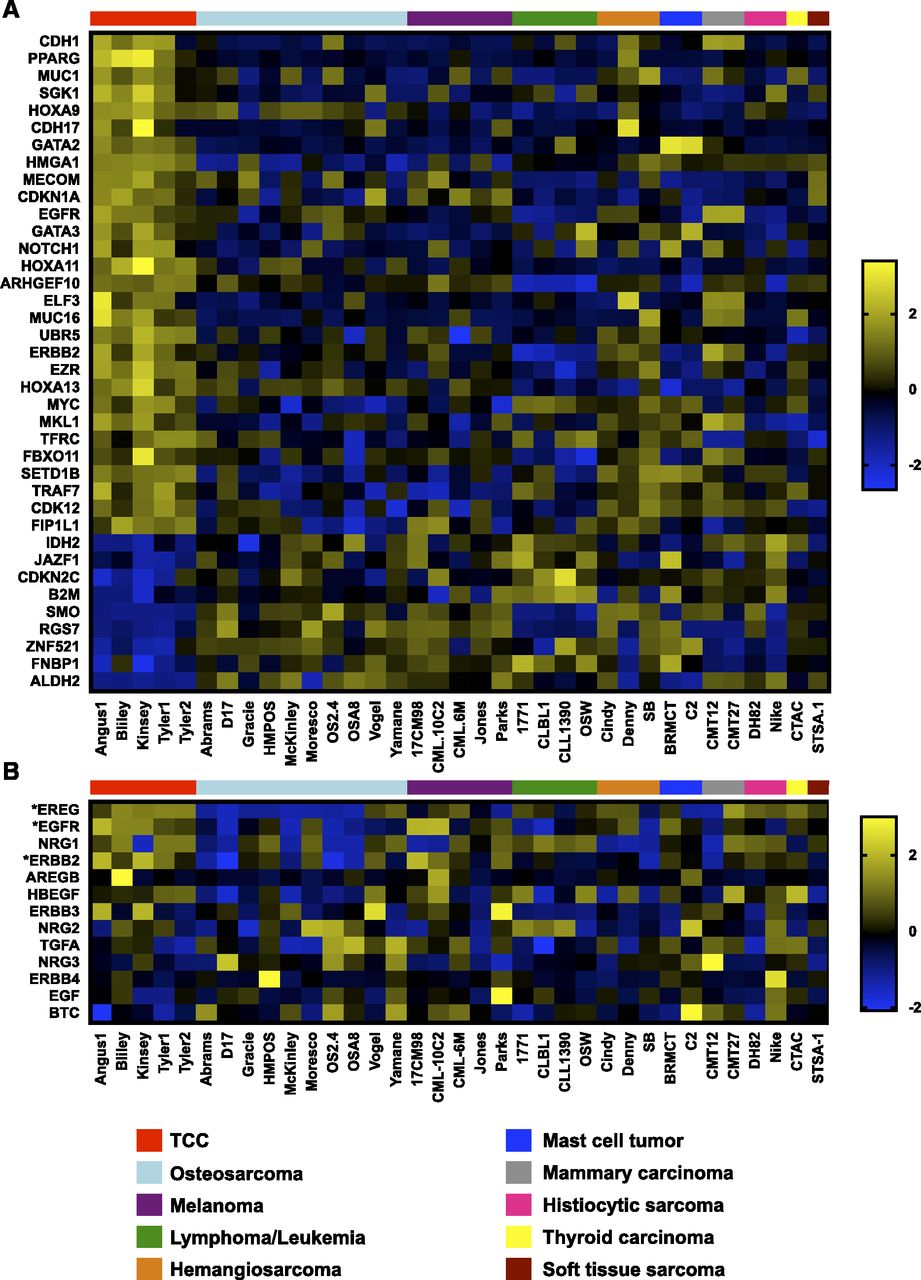
Twenty-nine and nine cancer genes were up- and downregulated in TCC cell lines relative to other cancer cell lines, respectively (Fig. 4A; Supplemental Table 2). This analysis revealed upregulation of genes encoding the EGFR and receptor tyrosine-protein kinase erbB-2 (ErbB2) receptors in TCC cell lines. We then expanded our analysis to all ErbB ligands and receptors present in the KEGG Pathway database (hsa04012, https://www.genome.jp/dbget-bin/www_bget?hsa04012) (Fig. 4B). In addition to upregulation of genes encoding EGFR and ErbB2 receptors, the ligand epiregulin was also upregulated in TCC cell lines. Additional cancer genes upregulated in TCC cell lines include CDH1, PPARG, NOTCH1, and MYC. Downregulated genes include IDH2, SMO, and ALDH2.

Upregulation of the ErbB signaling cascade in canine TCC cell lines. Microarray analysis was used to analyze gene expression in the FACC panel of canine cancer cell lines. (A) Cancer genes up- or downregulated in TCC relative to other canine cancer cell lines using a fold change cutoff of 1.5 and q < 0.05. (B) Expression of ErbB ligands and receptors (KEGG pathway: hsa04012) in FACC cell lines. Asterisks (*) indicate ErbB genes significantly upregulated in TCC cell lines. Colors on heat maps represent z-transformed expression values. Color bars above heat maps indicate cell line histotype.
Pan-ErbB Inhibition Synergizes with MAPK Inhibition in Canine TCC.
To determine whether ErbB inhibition alone or in combination with MAPK inhibition may be an effective therapy for canine TCC, TCC cell lines were treated with a pan-ErbB inhibitor sapitinib that targets EGFR, ErbB2, and ErbB3 receptors. All TCC cell lines had IC50 values greater than 1 μM (Supplemental Fig. 6). Combination treatments were performed to assess the efficacy of pan-ErbB inhibition with MAPK inhibition. Sapitinib treatment synergized with the paradox-breaking BRAFV600E inhibitor PLX7904 in BRAF mutant Bliley cells but not in wild-type Kinsey cells (median combination index of 0.4 vs. 1.4, respectively) (Fig. 5, A and B). Conversely, pan-ErbB inhibition synergized with MEK inhibition in both Bliley and Kinsey cells (median combination index of 0.7 and 0.5, respectively) (Fig. 5, C and D).

Combined MAPK and ErbB inhibition synergizes in canine TCC cell lines. Cells were treated with serial dilutions of the pan-ErbB inhibitor sapitinib (ERBBi) and (A and B) BRAF inhibitor PLX7904 (BRAFi) or (C and D) MEK1/2 inhibitor trametinib (MEKi), and cell proliferation was monitored for 72 hours. Drug synergy was determined using CalcuSyn software. (A and C) Colors on heat maps represent combination index (CI) values, where CI < 1 is synergistic (green), CI = 1 is additive, and CI > 1 is antagonistic (red). CI values were determined from three independent experiments (n = 3). (B and D) Representative images of red-labeled TCC cells 72 hours post-treatment. Images were acquired using the IncuCyte ZOOM Live-Cell Analysis System at 10× magnification. Scale bar, 300 μm.
Discussion
Recent studies have identified BRAF mutations in approximately 70% of canine TCC tumors (Decker et al., 2015; Mochizuki et al., 2015). Despite this discovery, little is known about the role that BRAF mutations play in canine TCC development and whether targeting mutant BRAF is a feasible therapy for TCC. In this study, we targeted BRAF and its downstream kinase, MEK, in five TCC cell lines: three BRAF mutants, one KRAS mutant, and one BRAF/KRAS wild-type. MAPK inhibitors suppressed proliferation in TCC cell lines with varying degrees of efficacy, but failed to sustain attenuation of MAPK pathway activity. The ErbB family of receptors was identified as a potential therapeutic target for TCC treatment, and inhibition of ErbB receptors synergized with MAPK inhibition in TCC cell lines. These data demonstrate the potential of ErbB receptor inhibition combined with either BRAF or MEK inhibition as an effective therapy for canine TCC. Additionally, our findings illustrate canine TCC’s potential utility as a naturally occurring model for investigating intrinsic resistance mechanisms to MAPK inhibition in human cancers and tailoring treatments to combat the emergence of resistance.
BRAF and KRAS mutant cell lines exhibited constitutive MAPK pathway activity based on expression levels of MAPK target genes (MPASs) and sustained ERK1/2 phosphorylation in the absence of FBS. Additionally, the Kinsey cell line, with no known MAPK mutation, also exhibited constitutive pathway activity. This phenomenon occurs in human cancers where ERK1/2 phosphorylation and expression of MAPK target genes do not always correlate with RAS/BRAF mutation status (Houben et al., 2008; Levidou et al., 2012; Wagle et al., 2018). Another group previously analyzed five different canine TCC cell lines and also showed sustained ERK1/2 phosphorylation in the absence of FBS for all cell lines (Rathore and Cekanova, 2014). Thus, constitutive MAPK activity seems to be a common occurrence in canine TCC, suggesting a possible causative role for the MAPK pathway in canine TCC pathogenesis. In human bladder cancer, BRAF mutations are rare; however, mutations in NRAS/HRAS occur in 6% of tumors and alterations in the RTK/Ras/PI3K pathway are present in 72% of tumors (Cancer Genome Atlas Research Network, 2014; Robertson et al., 2017).
All five TCC cell lines were insensitive to BRAF inhibition with vemurafenib relative to human BRAF mutant melanoma (A375) and colorectal (Colo205 and HT29) cell lines. In fact, TCC cell lines exhibited IC50 values similar to that of the RKO colorectal cancer cell line, which was previously reported to be insensitive to vemurafenib (Yang et al., 2012). Vemurafenib treatment yielded an initial decrease in MAPK pathway activity in BRAF mutant TCC cell lines, but pathway activity rebounded by 24 hours post-treatment. Human colorectal cancer cell lines also achieve MAPK pathway reactivation following 24 hours of vemurafenib treatment (Corcoran et al., 2012; Yang et al., 2012). Unlike canine TCC and human colorectal cancer cell lines, vemurafenib treatment in human melanoma cell lines maintains suppression of MAPK activation after 24 hours (Corcoran et al., 2012). Vemurafenib induced the same paradoxical MAPK activation in BRAF wild-type TCC cell lines that has been described in human BRAF wild-type cell lines. The mechanism behind paradoxical activation involves increased transactivation of RAF homo- or heterodimers as a result of vemurafenib binding, causing increased pathway activity (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010).
In melanoma patients, decreased BRAF allelic frequency is associated with a poorer clinical outcome to BRAF inhibition and combined BRAF/MEK inhibition (Lebbé et al., 2014; Stagni et al., 2018). The proposed mechanism behind this response is paradoxical MAPK pathway activation due to a higher wild-type allele frequency. BRAF mutations in the canine TCC cell lines used in this study are heterozygous; thus, we hypothesized that their reduced sensitivity to vemurafenib may be the result of paradoxical activation of the MAPK pathway due to the wild-type copy of BRAF. In support of this hypothesis, when treated with the paradox-breaking inhibitor PLX7904 BRAF mutant TCC cell lines were equally as sensitive as BRAF mutant human lines. Further studies in canine and human BRAF mutant cell lines are required to establish the role of BRAF zygosity in sensitivity to BRAF inhibitors.
In this study, canine TCC cell lines were equally sensitive to MEK1/2 inhibition with selumetinib or trametinib compared with BRAF mutant human cell lines. Our group assessed trametinib sensitivity for the entire Flint Animal Cancer Center panel of canine cancer cell lines and found that TCC cell lines were more sensitive than other cancer cell types (Das S, Idate R, Cronise KE, Gustafson DL, and Duval DL, unpublished data). Analysis of ERK1/2 activation following MEK1/2 inhibition for 6 hours showed complete or reduced pathway inhibition with trametinib and selumetinib, respectively. However, similar to the response with BRAF inhibition, TCC cell lines displayed a rebound in pathway activity by 24 hours post-treatment. Collectively, these data suggest canine TCC’s initial dependence on the RAS-BRAF-MEK signaling axis; however, intrinsic resistance mechanisms are able to bypass pathway inhibition by 24 hours.
In metastatic melanoma patients, tumor regression in response to BRAF inhibition correlates with sustained inhibition of ERK1/2 phosphorylation. Additionally, patients experiencing tumor regression typically had at least 80% inhibition of ERK1/2 phosphorylation following treatment (Bollag et al., 2010). This finding likely explains the lack of success of MAPK inhibition as a monotherapy for colorectal cancer treatment (Kopetz et al., 2015). Similar to the response of colorectal cancer cell lines (Corcoran et al., 2012), the TCC cell lines analyzed in this study showed reactivation of the MAPK pathway by 24 hours following MAPK inhibition, suggesting that long-term treatment with a MAPK inhibitor alone may not be an effective therapy for canine TCC.
Human melanoma and colorectal cancer exhibit acquired and innate resistance to BRAF inhibition, respectively. Mechanisms of acquired resistance in melanoma include BRAF splice isoforms, BRAF amplification, secondary NRAS mutations, CRAF overexpression, MEK1/2 mutations, and increased signaling through RTKs such as IGF-1R (Montagut et al., 2008; Nazarian et al., 2010; Villanueva et al., 2010; Poulikakos et al., 2011; Wagle et al., 2011; Shi et al., 2012). Mechanisms of intrinsic resistance in colorectal cancer include activation of RTKs (EGFR) and increased signaling through the PI3K/protein kinase B pathway (Corcoran et al., 2012; Prahallad et al., 2012; Mao et al., 2013). The majority of these resistance mechanisms involve reactivation of the MAPK pathway, explaining the increased efficacy in melanoma and colorectal cancer patients with dual inhibition of BRAF and MEK compared with monotherapy (Corcoran et al., 2015; Robert et al., 2015). A similar response was observed in this study where combined inhibition of BRAF and MEK was synergistic in the BRAF mutant Bliley TCC cell line.
To identify potential mediators of resistance to MAPK inhibition in TCC, in addition to determining other drivers that may contribute to TCC pathogenesis, we determined cancer genes that are differentially expressed in TCC cell lines versus other canine cancer cell lines. Genes upregulated in TCC cell lines include CDH1, PPARG, EGFR, ERBB2, EREG, and MYC. EGFR and ERBB2 protein expression is upregulated in 50% and 8%–30% of muscle-invasive human bladder cancers, respectively (Knowles and Hurst, 2015). Additionally, copy number alterations in EGFR (11%), ERBB2 (7%), PPARG (17%), and MYC (13%) are common in human bladder cancer (Cancer Genome Atlas Research Network, 2014).
Of particular interest to us was the upregulation of genes encoding ErbB receptors, EGFR and ERBB2, and the ErbB ligand epiregulin. Previous studies in colorectal cancer cell lines have shown that increased signaling through EGFR and/or ERBB2 allows cells to bypass BRAF inhibition (Corcoran et al., 2012; Prahallad et al., 2012). The mechanism behind the efficacy of combined ErbB and BRAF inhibition in colorectal cancer cell lines involves negative feedback regulation of the MAPK pathway. Increased MAPK signaling results in increased expression of negative regulators such as DUSP and SPRY. DUSP proteins inactivate ERK1/2 and SPRY proteins inhibit RTK-mediated activation of RAS (Lake et al., 2016). BRAF inhibition results in decreased expression of these negative regulators, thus relieving negative feedback of the MAPK pathway. In colorectal cancer it was suggested that this decrease in negative feedback results in increased activation of the EGFR and ERBB2 RTKs, and subsequent increased MAPK signaling through CRAF (Corcoran et al., 2012; Prahallad et al., 2012).
Similar to the response in colorectal cancer cell lines, MAPK inhibition with either a BRAF or MEK inhibitor synergized with a pan-ErbB inhibitor targeting EGFR, ERBB2, and ERBB3. These results suggest that the ErbB family of receptors and ligands may facilitate resistance to MAPK inhibition in canine TCC. Overall, the findings in this study suggest that dogs with BRAF mutant TCC may benefit from combined treatment with a pan-ErbB inhibitor and a paradox-breaking BRAF inhibitor, while dogs with BRAF wild-type TCC may benefit from combined pan-ErbB and MEK inhibition. Clinical trials evaluating the efficacy of these combinations in dogs with TCC may inform treatment modalities in human MAPK-driven cancers.
Acknowledgments
We thank Deanna Dailey for isolating total RNA from the Flint Animal Cancer Center cell lines and Rupa Idate for cell line validation.
Authorship Contributions
Participated in research design: Cronise, Hernandez, Gustafson, Duval.
Conducted experiments: Cronise, Hernandez.
Contributed new reagents or analytic tools: Gustafson.
Performed data analysis: Cronise, Hernandez, Duval.
Wrote or contributed to the writing of the manuscript: Cronise, Gustafson, Duval.
Footnotes
- Received January 15, 2019.
- Accepted April 27, 2019.
Funding was provided by the Morris Animal Foundation [D16CA-071] (to D.L.D.) and Shipley University Chair in Comparative Oncology (to D.L.G.).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal-regulated kinase
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase kinase
- MPAS
- mitogen-activated protein kinase pathway activity score
- PCR
- polymerase chain reaction
- PI3K
- phosphatidylinositol 3-kinase
- PLX7904
- 5-(2-cyclopropylpyrimidin-5-yl)-3-[3-[[ethyl(methyl)sulfamoyl]amino]-2,6-difluoro-benzoyl]-1H-pyrrolo[2,3-b]pyridine
- 5′ RACE
- 5′ rapid amplification of cDNA ends
- RTK
- receptor tyrosine kinase
- TCC
- transitional cell carcinoma
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}