


Abstract
Positive allosteric modulators (PAMs) of metabotropic glutamate receptor subtype 5 (mGlu5) have emerged as an exciting new approach for the treatment of schizophrenia and other central nervous system (CNS) disorders. Of interest, some mGlu5 PAMs act as pure PAMs, only potentiating mGlu5 responses to glutamate whereas others [allosteric agonists coupled with PAM activity (ago-PAMs)] potentiate responses to glutamate and have intrinsic allosteric agonist activity in mGlu5-expressing cell lines. All mGlu5 PAMs previously shown to have efficacy in animal models act as ago-PAMs in cell lines, raising the possibility that allosteric agonist activity is critical for in vivo efficacy. We have now optimized novel mGlu5 pure PAMs that are devoid of detectable agonist activity and structurally related mGlu5 ago-PAMs that activate mGlu5 alone in cell lines. Studies of mGlu5 PAMs in cell lines revealed that ago-PAM activity is dependent on levels of mGlu5 receptor expression in human embryonic kidney 293 cells, whereas PAM potency is relatively unaffected by levels of receptor expression. Furthermore, ago-PAMs have no agonist activity in the native systems tested, including cortical astrocytes and subthalamic nucleus neurons and in measures of long-term depression at the hippocampal Schaffer collateral-CA1 synapse. Finally, studies with pure PAMs and ago-PAMs chemically optimized to provide comparable CNS exposure revealed that both classes of mGlu5 PAMs have similar efficacy in a rodent model predictive of antipsychotic activity. These data suggest that the level of receptor expression influences the ability of mGlu5 PAMs to act as allosteric agonists in vitro and that ago-PAM activity observed in cell-based assays may not be important for in vivo efficacy.
Introduction
Metabotropic glutamate receptors (mGlus) are G protein-coupled receptors consisting of eight subtypes, termed mGlu1–8. mGlu receptors participate in a wide range of functions throughout the central nervous system (CNS) and are thought to play roles in multiple disease states (Niswender and Conn, 2010). In recent years, one mGlu receptor subtype, metabotropic glutamate receptor subtype 5 (mGlu5), has emerged as a novel target for treatment of schizophrenia and other disorders involving cognitive deficits. Current evidence suggests that schizophrenia may involve changes in activity through specific brain circuits involving glutamatergic transmission (Lisman et al., 2008; Marek et al., 2010) and that reduced activity of the N-methyl-d-aspartate (NMDA) subtype of ionotropic glutamate receptor may induce circuit changes similar to those observed in patients with schizophrenia. In contrast, it has been postulated that increased NMDA receptor activation could reduce symptoms observed in patients with schizophrenia (Ghoneim et al., 1985; Krystal et al., 1994). However, direct targeting of NMDA receptors is not practical because of the potential toxicity that can occur with overactivation of these ion channels (Hirose and Chan, 1993). Of interest, mGlu5 and the NMDA receptor are closely related signaling partners and activation of mGlu5 potentiates NMDA receptor signaling in multiple brain circuits (Awad et al., 2000; Doherty et al., 2000; Mannaioni et al., 2001; O'Brien et al., 2004). On the basis of this observation, selective activation of mGlu5 may provide an approach to compensate for the circuitry disruption that occurs in schizophrenia and similar disorders without inducing the adverse effects that occur with direct NMDA receptor activation.
Unfortunately, the orthosteric binding site of mGlu receptors is highly conserved across all eight mGlu receptor subtypes, making it difficult to develop selective mGlu5 agonists (Conn and Pin, 1997). A major breakthrough came with discovery of mGlu5-selective positive allosteric modulators (PAMs) that bind to a site on mGlu5 that is spatially removed from the orthosteric glutamate site (Conn et al., 2009). These novel mGlu5 PAMs provide high selectivity for mGlu5 relative to that of other mGlu receptor subtypes and have efficacy in a number of animal models used to predict antipsychotic and cognition-enhancing effects (for review, see Vinson and Conn, 2011).
A broad range of mGlu5 PAMs that induce robust increases in responses of mGlu5 to glutamate have been identified (O'Brien et al., 2004; Kinney et al., 2005; Chen et al., 2007; Liu et al., 2008; Rodriguez et al., 2010; Varnes et al., 2011). Of interest, mGlu5 PAMs can differ in ways that could have an important impact on their overall physiological and behavioral effects. For instance, some mGlu5 PAMs behave as pure PAMs and have no intrinsic agonist activity (i.e., no activity in the absence of the orthosteric agonist) when assessed using in vitro cell line assays, whereas others can serve as ago-PAMs to both potentiate responses to glutamate and directly activate mGlu5 when added in the absence of orthosteric agonists. At present, the functional relevance of pure PAM versus ago-PAM activity is not known. It has been suggested that pure PAMs may be preferred to ago-PAMs in that the requirement for activation of mGlu5 by endogenous glutamate would maintain normal patterns of receptor activity in response to neurotransmitter released from presynaptic terminals (Conn et al., 2009). However, all mGlu5 PAMs that have been previously optimized for in vivo use have weak intrinsic agonist activity (Kinney et al., 2005; Liu et al., 2008; Schlumberger et al., 2009; Rodriguez et al., 2010); therefore, it is possible that this ago-PAM activity is critical for efficacy in animal models.
To directly determine the potential relevance of pure PAM versus ago-PAM activity in native systems, we chemically optimized mGlu5 ago-PAMs that have maximal intrinsic agonist activity in cell lines along with close structural analogs that act as pure mGlu5 PAMs and are devoid of intrinsic agonist activity. Maintaining these activity profiles while optimizing for appropriate pharmacokinetic profiles and brain penetration provided unique tools that allowed studies of the functional relevance of ago-PAM versus pure PAM activity. Using these compounds with cell lines engineered to have different levels of mGlu5 expression, we found that ago-PAM activity is only seen in cell lines with high levels of mGlu5 expression. Furthermore, all compounds examined behaved as pure PAMs in native systems studied, regardless of whether they exhibited agonist activity in overexpressing cell lines. Finally, pure PAMs and ago-PAMs showed identical efficacy in reversing amphetamine-induced hyperlocomotor activity, a model used to predict potential antipsychotic activity. Together, these data suggest that the presence of ago-PAM activity in mGlu5-expressing cell lines is not a major determinant of activity in the systems studied and that pure PAMs and ago-PAMs have similar physiological and behavioral effects.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and antibiotics were purchased from Invitrogen (Carlsbad, CA). Dihydroxyphenylglycine (DHPG) was obtained from Ascent Scientific (Bristol, UK). N-Cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide (VU0360172), (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(4-hydroxypiperidin-1-yl)methanone (VU0361747), (4-hydroxypiperidin-1-yl)(4-phenylethynyl)phenyl)methanone (VU0092273) (Rodriguez et al., 2010), 6-(2-phenylethynyl)-1,2,3,4-tetrahydroisoquinolin-1-one (VU0240382) (Williams et al., 2011), and 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP) were synthesized in-house. Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of analytical grade.
Cell Culture
Two different human embryonic kidney (HEK) cell lines stably expressing rat mGlu5 (high and low expression) were maintained in complete DMEM supplemented with 10% FBS, 2 mM l-glutamine, 20 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, antibiotic/antimycotic, and G418 (500 μg/ml; Mediatech, Herndon, VA) at 37°C in a humidified incubator containing 5% CO2. HEK293 cells stably expressing rat mGlu1 were maintained in the same media as the mGlu5 cells. HEK293 cells stably expressing G protein-coupled inwardly rectifying potassium channels (HEK293-GIRK) along with the individual group II and group III mGlu receptors were maintained in growth media containing 45% DMEM, 45% F-12, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, antibiotic/antimycotic, nonessential amino acids, G418 (700 μg/ml), and puromycin (0.6 μg/ml). HEK cells expressing human mGlu5 under the control of a tetracycline promoter were maintained in the same media as the rat mGlu5 cells except that blasticidin (5 μg/ml) and phleomycin (Zeocin; Invitrogen) (150 μg/ml) were substituted for G418. These cells were treated with increasing concentrations of tetracycline hydrochloride (1–10 ng/ml) in media for 22 h to induce receptor expression before their use in fluorescence-based calcium assays.
Fluorescence-Based Calcium Assays
For measurement of compound-evoked increases in intracellular calcium, HEK293 cells stably expressing rat mGlu5 were plated in 96-well, poly-d-lysine-coated, black-walled, clear-bottomed plates (Greiner Bio-One, Monroe, NC) in 100 μl of assay medium (DMEM supplemented with 10% dialyzed fetal bovine serum, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 4 to 5 × 104 cells/well. Cells were grown overnight at 37°C and 5% CO2. On the next day, medium was removed from the cells, and they were incubated with 75 μl/well of 2 μM Fluo-4AM (Invitrogen) prepared as a 2.3 mM stock solution in dimethyl sulfoxide (DMSO), mixed in a 1:1 ratio with 10% (w/v) Pluronic acid F-127, and diluted in calcium assay buffer [Hank's balanced salt solution (HBSS) (Invitrogen) supplemented with 20 mM HEPES and 2.5 mM probenecid, pH 7.4] for 1 h at 37°C. Dye loading solution was removed and replaced with 90 μl/well of assay buffer. For PAM potency curves, mGlu5 compounds were diluted at a 3× concentration and added to the cells at 20 s followed by the addition of a 10× concentration of an EC20 of glutamate or DHPG at 80 s into a 140-s protocol. The EC20 concentration was determined daily because of some variability (glutamate, high expression 150–200 nM and low expression 40–50 nM; DHPG, high expression 500 nM). For fold shift experiments, multiple fixed concentrations of mGlu5 compound (final concentrations of 30 nM–30 μM) or vehicle were added (3× stock) at 20 s followed by the addition of a concentration-response curve (CRC) of glutamate (10× stock) at 80 s into a 140-s protocol. All experiments were matched in solvent (DMSO) concentration. Calcium flux was measured over time as an increase in fluorescence using a FlexStation II (Molecular Devices, Sunnyvale, CA) using an excitation wavelength of 488 nm, an emission wavelength of 525 nm, and a cutoff wavelength of 515 nm. The increase in fluorescence over basal was determined for the peak and plateau phases of the response before being normalized to the maximal peak response elicited by glutamate (10–100 μM). Data were transformed and fitted using Prism (GraphPad Software Inc., San Diego, CA).
Rat Cortical Astrocytes
Primary rat cortical astrocytes were received from Lonza (Basel, Switzerland) and stored in liquid nitrogen until use. Astrocytes were thawed following the protocol provided by Lonza and plated on BD Falcon Primaria dishes in assay growth media (AGM) (assay basal media supplemented with AGM SingleQuots from Lonza). Astrocytes were fed with AGM 4 to 5 h after initial plating and then every 3 to 4 days until confluent. Astrocytes were plated in 96-well, poly-d-lysine–coated, black-walled, clear-bottomed plates in 100 μl of AGM at a density of approximately 5 × 104 cells/well. On the next day astrocytes were supplemented with G5 diluted 1:100 in AGM. The calcium flux assay was performed on the following day using assay conditions and compound preparation identical to those used for the mGlu5 HEK293 cell assay.
Radioligand Binding
HEK293 cells expressing high or low levels of rat mGlu5 or rat cortical astrocytes were assayed to determine the level of mGlu5 receptor expression. Cells were harvested by trypsinization and pelleted by centrifugation for 3 min at 300g. Cells were resuspended in calcium assay buffer. For saturation binding experiments, cells were incubated with two concentrations of [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine (6 or 90 nM) for 1 h at room temperature in calcium assay buffer. The Kd of [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine for rat mGlu5 has been previously determined to be 5.7 nM. Nonspecific binding was determined using 10 μM 2-methyl-6-(phenylethynyl)pyridine hydrochloride. Binding assays were terminated by rapid filtration through GF/B Unifilter plates (PerkinElmer Life and Analytical Sciences, Waltham, MA) using a Brandel 96-well plate harvester (Brandel Inc., Gaithersburg, MD), followed by three washes with ice-cold (4°C) binding buffer (50 mM Tris-HCl and 0.9% NaCl, pH 7.4), separating bound from free radioligand. Plates were allowed to dry overnight before addition of MicroScint 20 (40 μl/well; PerkinElmer Life and Analytical Sciences). Radioactivity of bound radioligand was counted at least 2 h after addition of scintillation fluid using a TopCount Scintillation Counter (PerkinElmer Life and Analytical Sciences).
Selectivity Screening
mGlu1.
HEK293 cells stably expressing rat mGlu1 were plated in 384-well, poly-d-lysine-coated, black-walled, clear-bottomed plates in 20 μl of assay medium at a density of 2 × 104 cells/well. Assays were performed on the following day using the High-Throughput Screening Center at Vanderbilt University. The medium was removed and replaced with assay buffer (HBSS, 20 mM HEPES, pH 7.4, and 2.5 mM probenecid) containing 1 μM Fluo4-AM using a BioTek ELx washer. The cells were incubated for 45 min at 37°C5% CO2 followed by a second ELx wash with assay buffer containing no dye. After a 10-min equilibration period, cell plates were introduced into the Functional Drug Screening System 6000 (Hamamatsu, Hamamatsu City, Japan) for calcium flux measurements. To assess the effect of the modulator, either vehicle or a fixed concentration of test compound (10 μM) was added followed 140 s later by a concentration-response curve of glutamate. The change in relative fluorescence over basal was calculated before normalization to the maximal response to glutamate.
Group II and Group III mGlus.
The functional activity of the compounds of interest was assessed at the rat group II and III mGlu receptors by measuring thallium flux through GIRK channels as described in detail previously (Niswender et al., 2008). In brief, HEK293-GIRK cells expressing mGlu subtype 2, 3, 4, 6, 7, or 8 were plated into 384-well, poly-d-lysine-coated, black-walled, clear-bottomed plates at a density of 1.5 × 104 cells/well in 20 μl of assay medium. On the following day, the medium was removed and replaced with assay buffer (HBSS and 20 mM HEPES, pH 7.4) supplemented with 0.16 μM FluoZin-2 AM (Invitrogen). (See methods above for further details.) A single concentration of test compound (10 μM) or vehicle was added followed 140 s later by a concentration-response curve of glutamate [or l-(+)-2-amino-4-phosphonobutyric acid (l-AP4) for mGlu7] diluted in thallium buffer (125 mM NaHCO3, 1 mM MgSO4, 1.8 mM CaSO4, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES), and fluorescence was measured using a Functional Drug Screening System 6000. Data were analyzed as described previously (Niswender et al., 2008).
Brain Slice Electrophysiology
Extracellular Field Potential Recordings.
All animals used in these studies were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). Young adult (5- to 6-week-old) male Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) were anesthetized with isoflurane and decapitated, and the brains were quickly removed and submerged into ice-cold cutting solution [110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 5 mM d-glucose, 0.6 mM (+)-sodium-l-ascorbate, 0.5 mM CaCl2, and 7 mM MgCl2] continuously bubbled with 95% O2/5% CO2. The brains were then hemisected, and 400-μm transverse slices were made using a Vibratome (Leica VT100S; Leica Microsystems, Nussloch, Germany). Individual hippocampi were microdissected from the slice and transferred to a room temperature mixture containing equal volumes of cutting solution and artificial cerebrospinal fluid (aCSF) (125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 2 mM CaCl2, and 1 mM MgCl2), where they were allowed to equilibrate for 30 min. The hippocampi were then transferred to aCSF continuously bubbled with 95% O2/5% CO2 for an additional hour. Slices were transferred to a submersion recording chamber and allowed to equilibrate for 5 to 10 min at 30–32°C with a flow rate of 2 ml/min. A bipolar-stimulating electrode was placed in the stratum radiatum near the CA3-CA1 border to stimulate the Schaffer collaterals. Recording electrodes were pulled with a Flaming/Brown micropipette puller (Sutter Instrument Company Novato, CA), filled with aCSF, and placed in the stratum radiatum of area CA1. Field potential recordings were acquired using a MultiClamp 700B amplifier (Molecular Devices) and pCLAMP 9.2 software (Molecular Devices). Input-output curves were generated to determine the stimulus intensity that produced 40 to 60% of the maximum field excitatory postsynaptic potential (fEPSP) slope before each experiment. This submaximal fEPSP slope was used as the baseline stimulation for the remainder of the individual experiment. mGlu5 compounds were diluted to the appropriate concentrations in DMSO (0.1%) in aCSF and applied to the bath for 10 to 20 min using a perfusion system. Chemically induced mGlu long-term depression (LTD) was initiated by the application of DHPG in aCSF (25–75 μM) for 10 min. Sampled data were analyzed offline using Clampfit 9.2 (Molecular Devices). Three sequential fEPSPs were averaged, and their slopes were calculated. All fEPSP slopes were normalized to the average slope calculated during the predrug period (percentage of baseline). Data were analyzed using GraphPad Prism.
Whole-Cell Patch-Clamp Recordings.
Whole-cell patch-clamp recordings were performed using midbrain slices prepared from 15- to 19-day-old Sprague-Dawley rats. After decapitation, brains were removed quickly and submerged into ice-cold cutting solution (200 mM sucrose, 1.9 mM KCl, 1.2 mM NaH2PO4, 25 mM NaHCO3, 10 mM d-glucose, 0.5 mM CaCl2, and 6 mM MgCl2) and equilibrated with 95% O2/5% CO2. Brains were hemisected, and sagittal brain slices containing the subthalamic nucleus (STN) were cut at 300 μm using a Vibratome. Slices were transferred to a holding chamber containing aCSF (125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM d-glucose, 2 mM CaCl2, and 1 mM MgCl2) supplemented with 5 μM glutathione to increase slice viability, continuously bubbled with 95% O2/5% CO2 at 32°C, and incubated for 30 min. Slices were then maintained at room temperature in aCSF equilibrated with 95% O2/5% CO2 until they were transferred to the submersion recording chamber and perfused with room temperature aCSF at a rate of 2 ml/min. The same protocol was used for striatal slices except that slices were cut coronally. Neurons in the STN or striatum were visualized with a 40× water immersion lens with Hoffman modulation contrast optics coupled with an Olympus BX50WI upright microscope (Olympus America Inc., Center Valley, PA). Borosilicate glass patch electrodes were pulled using a Flaming/Brown micropipette puller with a resistance of 2 to 5 MΩ when filled with an intracellular solution (123 mM potassium gluconate, 7 mM KCl, 0.025 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 0.1 mM EGTA, 2 mM sodium-ATP, and 0.2 mM sodium-GTP; pH adjusted to 7.3 with 1 N KOH; osmolarity, 290 mOsM). All whole-cell patch-clamp recordings were performed using a MultiClamp 700B amplifier. Data were digitized with a DigiData 1331 system (Molecular Devices), filtered at 2 kHz, and acquired using pCLAMP 9.2. After formation of a whole-cell configuration, cells were current-clamped to −60 mV for STN neurons; medium spiny neurons were maintained at their normal resting potential (approximately −75 mV), and changes in membrane potential were recorded. Data were analyzed using Clampfit 9.2.
Brain Homogenate Binding
The brain homogenate binding of each compound was determined in rat brain homogenates via equilibrium dialysis using RED plates (Thermo Fisher Scientific, Waltham, MA) as described previously (Wan et al., 2007). In brief, brain tissue homogenate from rats was prepared by diluting 1 volume of whole-brain tissue with 3 volumes of phosphate buffer (25 mM, pH 7.4). The mixture was then subjected to mechanical homogenization using a Mini-Beadbeater and 1.0-mm Zirconia/Silica Beads (BioSpec Products, Bartlesville, OK). Brain homogenate (220 μl) was added to the 96-well plate containing the test compound (5 μl) and mixed thoroughly for a final concentration (5 μM) of test compound. Then, 200 μl of the brain homogenate-compound mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μl of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated for 4 h at 37°C with shaking. At completion, 50-μl aliquots from each chamber were diluted 1:1 (50 μl) with either brain homogenate (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile containing 50 nM carbamazepine as an internal standard was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min), and supernatants were transferred and diluted 1:1 (supernatant/water) into a new 96-well plate, which was then sealed in preparation for liquid chromatography-tandem mass spectrometry analysis. Each compound was assayed in triplicate within the same 96-well plate. Brain homogenate binding samples were analyzed using liquid chromatography-tandem mass spectrometry techniques on a Thermo Fisher Scientific TSQ Quantum Ultra triple quad detector via electrospray ionization with two Thermo Fisher Scientific Accela pumps and a CTC PAL autosampler (Leap Technologies, Carrboro, NC). Analytes were separated by gradient elution on a dual-column system with two Acquity BEH C18, 2.1 × 50 mm, 1.7-μm columns (Waters, Milford, MA) heated at 40°C. HPLC mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. Pump 1 runs the gradient: 90:10 (A/B) at 800 μl/min hold 0 to 0.3 min, linear ramp to 5:95 (A/B) 0.3 to 0.8 min, 5:95 (A/B) hold 0.8 to 1.2 min, and return to 90:10 (A/B) at 1.3 min. While pump 1 runs the gradient method, pump 2 equilibrates the other column isocratically with 90:10 (A/B). The total run time is 1.5 min/injection. All compounds were optimized using Thermo Fisher Scientific QuickQuan software:
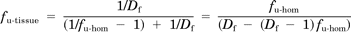 where fu-hom and Df represent the measured fraction unbound in dilute homogenate and the dilution factor, respectively (Wan et al., 2007).
where fu-hom and Df represent the measured fraction unbound in dilute homogenate and the dilution factor, respectively (Wan et al., 2007).
A critical question for optimizing novel mGlu5 PAMs as potential therapeutic agents or in vivo tools is whether ago-PAM activity is important for in vivo efficacy. To address this important question, it is essential to achieve adequate pharmacokinetic and physiochemical properties that provide sufficient CNS exposure to assess in vivo efficacy after systemic dosing. In addition to optimizing these mGlu5 PAMs to achieve pure PAM versus ago-PAM activity, chemistry efforts were also made to optimize the molecules to achieve high CNS exposure for representatives of each class. The free fraction of novel CNS-active compounds in the brain is a critical parameter for achieving high brain exposure. In general, we set a criterion of achieving at least 1% free fraction in assays of brain homogenate binding before advancing to in vivo pharmacokinetic studies. Incubation of compound with brain homogenate samples resulted in a free fraction of 2.9% for VU0360172, 9.6% for VU0361747 and 2.5% for VU0092273 (Supplemental Fig. 1). In contrast, the free fraction of VU0240382 was very limited at 0.01%, which prohibited it from being assessed further for use in behavioral studies (Supplemental Fig. 1).
In Vivo Pharmacokinetic Analysis
Test compound was formulated as 10% Tween 80 in sterile water at the concentration of 3.33 mg/ml and administered intraperitoneally to male Sprague-Dawley rats weighing 275 to 300 g (Harlan, Indianapolis, IN) at a dose of 10 mg/kg. Rat blood (cardiac puncture) and brain were collected at 0.25, 0.5, 1, 3, and 6 h. Animals were euthanized and decapitated, and the brains were removed, thoroughly washed in ice-cold (4°C) phosphate-buffered saline, and immediately frozen on dry ice. Plasma was separated by centrifugation and stored at −80°C until analysis. On the day of analysis, frozen whole rat brains were weighed and diluted with 1:3 (w/w) parts of 70:30 isopropanol/water. The mixture was then subjected to mechanical homogenization using a Mini-Beadbeater and 1.0-mm Zirconia/Silica Beads. The sample extraction of plasma (20 μl) and brain homogenate (20 μl) was performed by a method based on protein precipitation using 3 volumes of ice-cold acetonitrile containing an internal standard (carbamazepine) with a final concentration of 50 ng/ml. Extracts were centrifuged at 4000g for 5 min. The supernatants of plasma and brain homogenate extracts were then diluted 1:1 with water.
Samples were analyzed via electrospray ionization on an API 4000 (AB Sciex, Foster City, CA) triple quadrupole instrument that was coupled with LC-10AD pumps (Shimadzu, Columbia, MD) and a CTC PAL autosampler. Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3-μm column (Fortis Technologies Ltd., Cheshire, UK) thermostated at 40°C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), and mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 10% B after a 0.2-min hold and was linearly increased to 90% B over 0.8 min; held at 90% B for 0.5 min, and returned to 30% B in 0.1 min followed by a reequilibration (0.9 min). The total run time was 2.5 min, and the HPLC flow rate was 0.5 ml/min. The source temperature was set at 50°C, and mass spectral analyses were performed using multiple reaction monitoring, with transitions specific for each compound using a TurboIonSpray source in positive ionization mode (5.0 kV spray voltage). The calibration curves were constructed, and linear response was obtained in the range of 20 to 10,000 ng/ml by spiking known amounts of VU0360172, VU0361747, or VU0092273 in blank brain homogenates and plasma. All data were analyzed using AB Sciex Analyst 1.5.1 software. The final pharmacokinetic parameters were calculated by noncompartmental analysis using WinNonlin software (version 5.1; Pharsight, Mountain View, CA).
In vivo pharmacokinetic analysis revealed that VU0360172, VU0361747, and VU0092273 demonstrated rapid and significant absorption after intraperitoneal dosing (Supplemental Fig. 1). Maximum plasma concentrations of 4450 ng/ml VU0360172 were observed at 0.5 h after administration. VU0361747 also reached peak plasma levels (4690 ng/ml) at 0.5 h, whereas VU0092273 obtained a maximum concentration of 4863 ng/ml at 1 h. All three allosteric modulators also had rapid and significant uptake into the brain after systemic administration (Supplemental Fig. 1). VU0360172 reached maximum levels of 2000 ng/g in whole-brain tissues with a tmax of 0.5 h and AUCbrain/AUCplasma ratio of 0.46. The tmax of VU0092273 was 0.5 h with a Cmax of 19,261 ng/g and AUCbrain/AUCplasma of 4.47. Similar to the tmax of plasma, VU0361747 reached maximum brain concentrations of 5739 ng/g at 0.5 h with 1.12 AUCbrain/AUCplasma. These pharmacokinetic properties, including brain/plasma ratios and acceptable free fractions, of each of these compounds support their use for in vivo behavioral testing and provide an optimal dose regimen for behavioral studies.
Amphetamine-Induced Hyperlocomotion
Subjects.
Studies were conducted using male Sprague-Dawley rats weighing 280 to 310 g. Subjects were housed three per cage under a 12-h light/dark cycle (lights on at 6:00 AM) with access to food and water ad libitum. Testing procedures were performed between 7:00 AM and 6:00 PM. Dose groups consisted of 5 to 12 rats per group. All test compounds were dissolved in 10% Tween 80 and double-deionized water, and the pH was adjusted to approximately 7.0 using 1 N NaOH. Test compounds were administered intraperitoneally at a dose of 56.6 mg/kg in a 3 ml/kg volume. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and were approved by the institutional animal care and use committee.
Apparatus.
A SmartFrame Open Field System (Kinder Scientific, San Diego, CA) equipped with 32 horizontal (x- and y-axes) infrared photobeams positioned 1 cm above the floor of the chamber was used to conduct the studies. Ambulation or locomotor activity was measured as the number of total photobeam breaks per 5-min interval and was recorded with a Pentium I computer equipped with Motor Monitor System software (Kinder Scientific).
Procedure.
Rats were placed in the open-field chambers and allowed to habituate for 30 min followed by intraperitoneal pretreatment with vehicle or test compound. After an additional 30 min, rats received a saline vehicle or 1 mg/kg amphetamine subcutaneous injection. Locomotor activity was measured for an additional 60 min.
Analysis.
The main effects of test compound treatment on the locomotor activity area under the time course curve were evaluated using one-way analysis of variance. Comparisons of treatment group effects relative to the vehicle plus amphetamine group were completed across the time interval from t = 60 to 120 min using Dunnett's post hoc tests. p < 0.0001 versus vehicle plus amphetamine group.
Results
Discovery of Structurally Related Pure mGlu5 PAMs and mGlu5 Ago-PAMs.
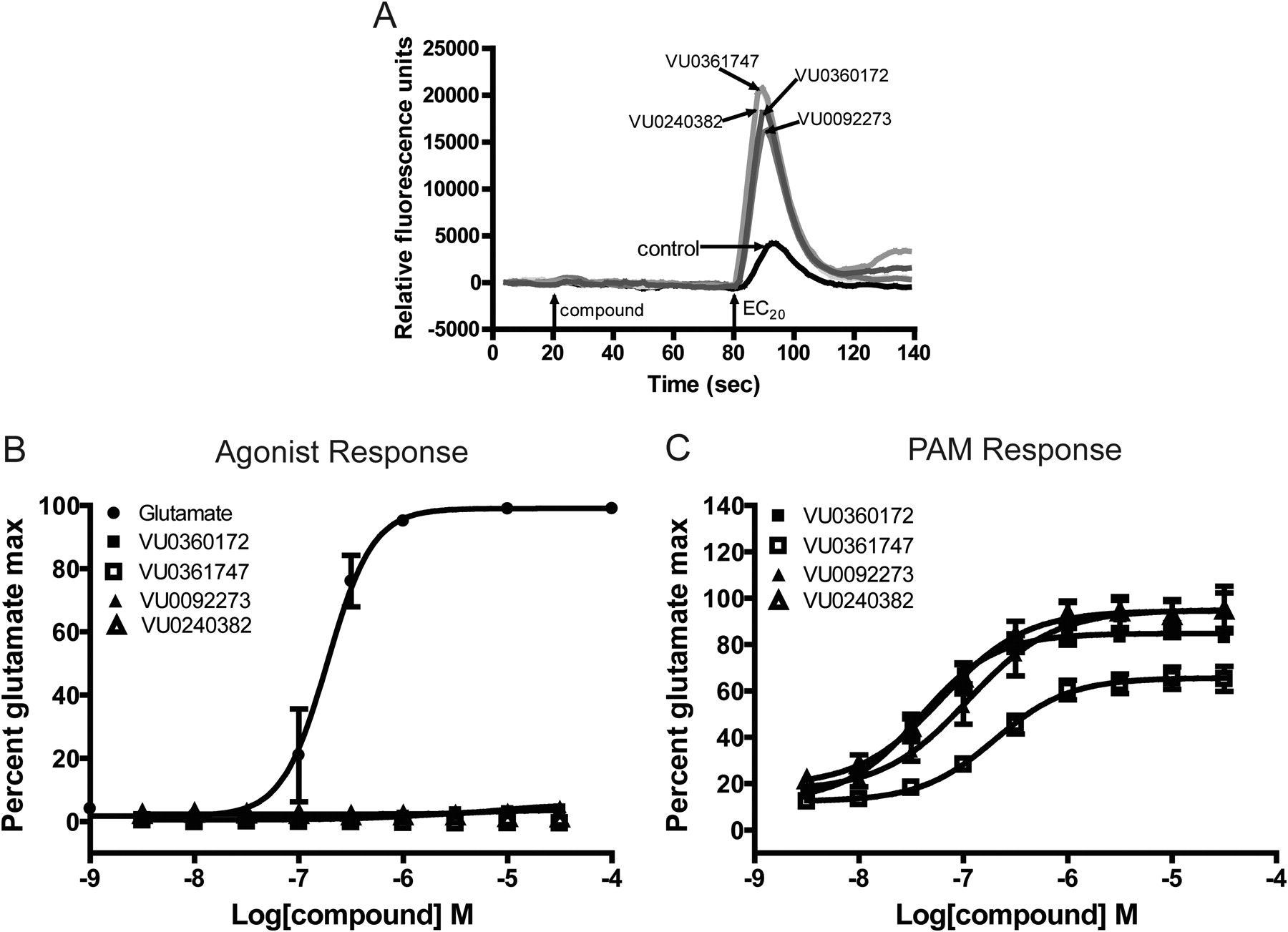
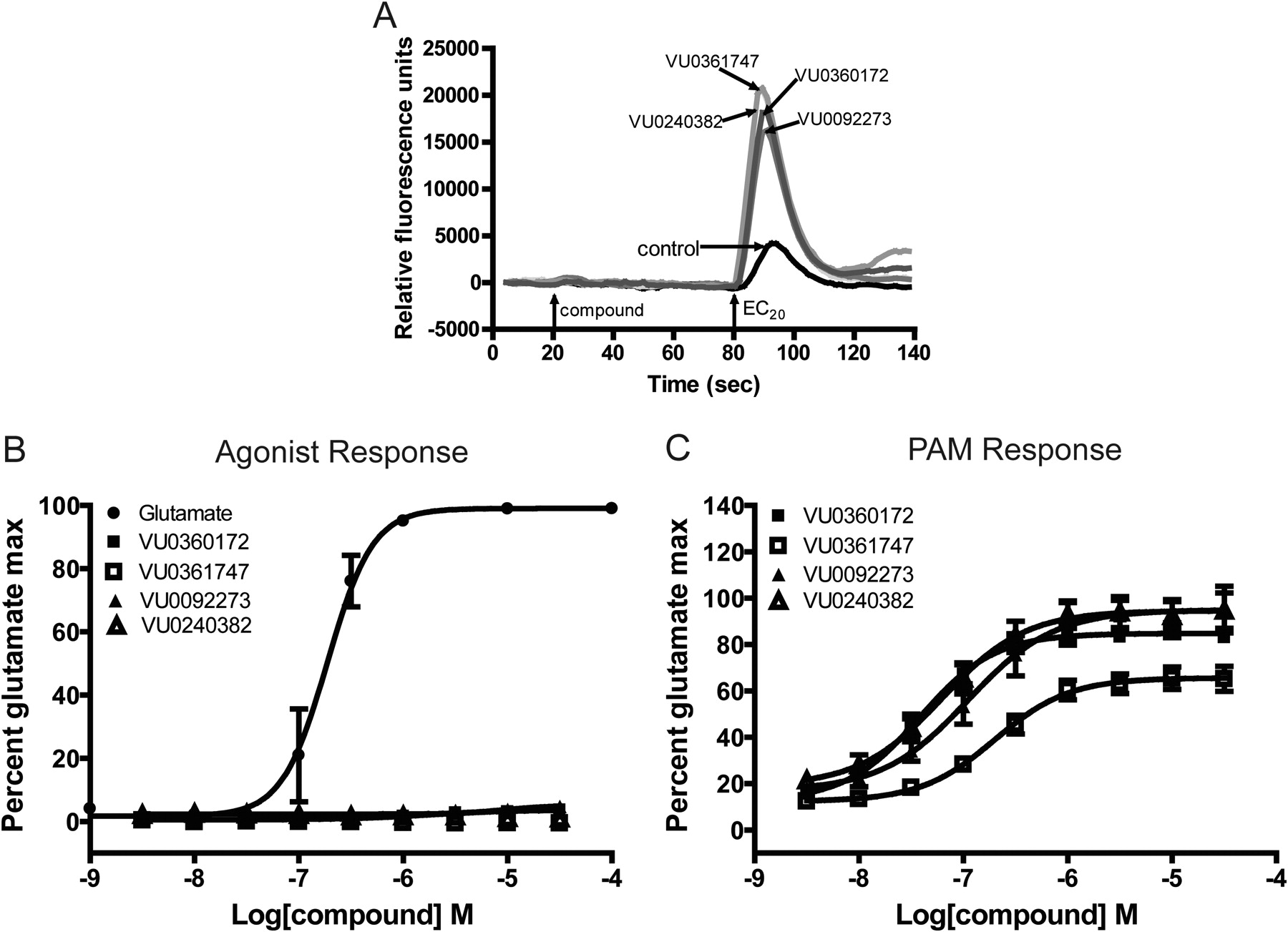
We recently identified a novel series of mGlu5 PAMs that are derived from a biaryl acetylene scaffold (Rodriguez et al., 2010; Williams et al., 2011) (Fig. 1). We chose to examine two scaffolds within the biaryl acetylene class based on the identity of the eastern aryl ring with the presence of either a phenyl or nicotinamide-based structure. In initial experiments, we used an established cell line that expresses a high density of mGlu5 (2.3 ± 0.4 pmol/mg) and has been used for discovery and extensive characterization of mGlu5 PAMs in our previous studies (Rodriguez et al., 2005, 2010; Chen et al., 2007; Williams et al., 2011). This cell line was used to support an extensive chemical optimization and pharmacological characterization effort in which we identified VU0360172 and VU0092273 as mGlu5 PAMs that exhibit robust ago-PAM activity. This profile can be seen in Fig. 2A where VU0360172 and VU0092273 (10 μM) induce strong agonist responses upon addition in the absence of glutamate and also potentiate the response to glutamate (EC20) that is added 60 s after addition of the test compound. We also identified two close structural analogs of VU0360172 and VU0092273 termed VU0361747 and VU0240382 (Fig. 1). In contrast to VU0360172 and VU0092273, VU0361747 and VU0240382 behaved as pure PAMs in vitro (Fig. 2B) and did not activate mGlu5 when added alone (10 μM) but potentiated the response to a subsequent addition of an EC20 concentration of glutamate (Fig. 2B). Figure 2C shows a full concentration-response relationship for agonist activity of each compound in the absence of glutamate addition. Consistent with the responses to addition of 10 μM concentrations of each PAM, VU0360172 and VU0092273 induced concentration-dependent activation of mGlu5 with potencies of 220 ± 25 nM and 1.3 ± 0.2 μM, respectively. In contrast, VU0361747 and VU0240382 induced little or no activation of mGlu5 at any concentration tested (Fig. 2C). We then established the potencies of each compound as a PAM by evaluating the effects of multiple concentrations of each compound in the presence of an EC20 concentration of glutamate (Fig. 2D). Each of the mGlu5 PAMs tested induced a concentration-dependent potentiation of the response to glutamate with EC50 values of 13 ± 2 nM (VU0360172), 35 ± 5 nM (VU0092273), 126 ± 23 nM (VU0361747), and 39 ± 10 nM (VU0240382) (mean ± S.E.M., n = 3–6). Although the agonist activity of these compounds has not been evaluated previously, these potencies for the PAM response agree with values that were reported previously by Rodriguez et al. (2010) for VU0360172, VU0092273, and VU0361747.
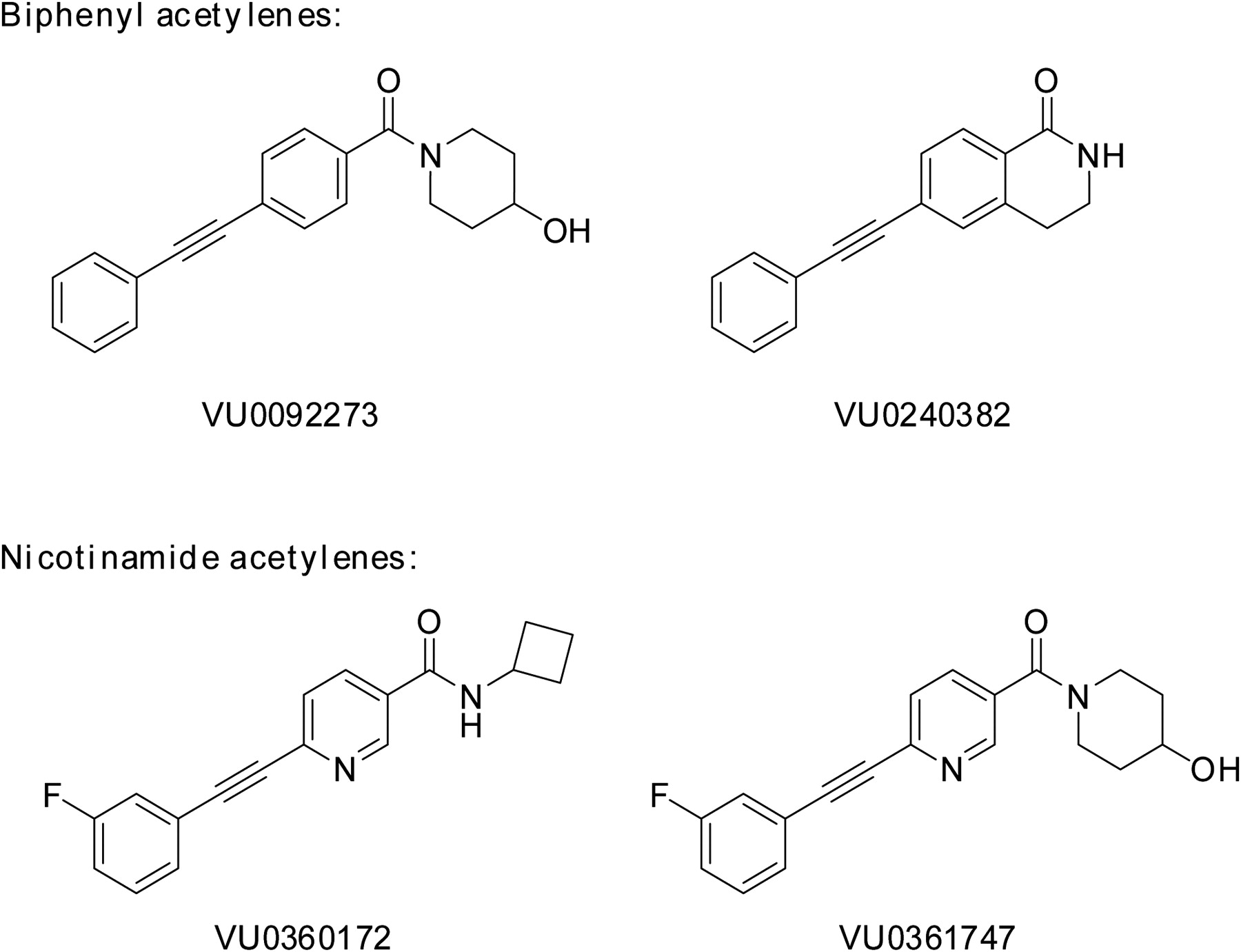
Structures of mGlu5 allosteric modulators used in this study. Biphenyl acetylenes: VU0092273 and VU0240382; nicotinamide acetylenes: VU0360172 and VU0361747.

mGlu5 allosteric modulators exhibit distinctions in vitro in their ability to modulate glutamate-induced calcium mobilization. A and B, representative raw calcium traces from one experiment show the effect of 10 μM mGlu5 modulators alone (compound; 20–80 s; agonist) and in response to an EC20 concentration of glutamate (80–140 s; PAM); the response to an EC20 concentration of glutamate in the presence of vehicle alone (control) is shown for comparison. A, compounds classified as ago-PAMs. B, compounds classified as pure PAMs. C and D, potencies of the compounds were assessed by adding a concentration-response curve of the mGlu5 compound to cells before the addition of an EC20 concentration of glutamate. The calcium response was measured and normalized to a maximally effective concentration of glutamate (100 μM). C, a concentration-dependent increase in agonist activity was observed for VU0360172 (■) and VU0092273 (▴). A glutamate CRC is included as a positive control (●). D, all compounds enhanced the response to an EC20 concentration of glutamate in a concentration-dependent manner. Data represent the mean ± S.E.M. of three to six independent experiments performed in duplicate.
mGlu5 Allosteric Modulators Are Selective for mGlu5.
Discovery of structurally related mGlu5 PAMs that have robust ago-PAM activity versus pure PAM activity could provide important new tools for understanding the functional impact of these two classes of compounds. However, to use these as tools in later physiological or behavioral assays, it is important to establish whether they are selective for mGlu5 relative to other mGlu receptor subtypes. We evaluated the effects of 10 μM concentrations of each compound on its ability to shift the glutamate CRC in calcium mobilization (mGlu1) or thallium flux (mGlu2, mGlu3, mGlu4, mGlu6, or mGlu8) assays using cell lines that have been extensively used to characterize the effects of allosteric modulators of each of the other mGlu receptor subtypes (Niswender et al., 2008; Rodriguez et al., 2010). For mGlu7, l-AP4 was used as the agonist in the thallium flux assay because of the low potency of glutamate at this mGlu receptor subtype. All of the compounds were inactive in shifting the concentration-response curves for glutamate in cells expressing other mGlu receptor subtypes (Supplemental Fig. 2). The one exception was VU0092273, which slightly shifted the glutamate CRC at mGlu3 to the right and downward, suggesting that VU0092273 may have weak antagonist activity at mGlu3. This is in agreement with the previously reported effects of VU0092273, including very weak inhibition of the mGlu3 response (Rodriguez et al., 2010).
Receptor Expression Level Influences the Shift from Pure PAM to Ago-PAM Activity.
We next assessed the influence of receptor expression level on the responses to these mGlu5 ago-PAMs and pure PAMs. To accomplish this, we developed a new HEK293 cell line that was selected for expression of lower levels of mGlu5. After extensive characterization of this cell line to assess mGlu5 density and to ensure appropriate responses to orthosteric ligands (data not shown), we compared the activity of these four compounds in our original cell line (mGlu5 Bmax = 25.2 ± 1.5 fmol/105 cells) with the phenotypes induced in a second cell line that was developed to provide lower mGlu5 expression (mGlu5 Bmax = 7.8 ± 0.6 fmol/105 cells). In contrast to the effects observed with the high expressing cell line, none of the compounds tested induced an agonist response in the cell line with a lower density of mGlu5 receptors (Fig. 3, A and B). However, glutamate induced a concentration-dependent activation of mGlu5 (Fig. 3B) with a potency similar to that observed in the higher expression cell lines [glutamate EC50 = 200 ± 42 nM (n = 4) versus 360 ± 70 nM (n = 6) in the high expression cell line] (Fig. 2C). Furthermore, all compounds potentiated the response to an EC20 concentration of glutamate (Fig. 3C) with potencies similar to those observed for potentiation of glutamate responses in the high expression line (EC50 values: VU0360172, 39 ± 5 nM; VU0092273, 122 ± 19 nM; VU0240382, 64 ± 10 nM; VU0361747, 200 ± 5 nM; mean ± S.E.M., n = 3–4). These data suggest that the ability of mGlu5 ago-PAMs to activate the receptor in the absence of an orthosteric agonist may be highly sensitive to levels of receptor expression.
In contrast to results seen with a cell line expressing a high density of mGlu5, mGlu5 allosteric modulators act exclusively as PAMs in a cell line with lower receptor expression. A, representative raw calcium traces from one experiment show that 10 μM mGlu5 modulator alone (compound; 20–80 s; agonist) induces no agonist response but potentiates an EC20 concentration of glutamate (80–140 s; PAM). The response to an EC20 concentration of glutamate in the presence of vehicle alone (control) is shown for comparison. B and C, potencies of mGlu5 allosteric modulators were determined by addition of a CRC of test compound followed by the addition of an EC20 concentration of glutamate. The calcium response was normalized to a maximally effective concentration of glutamate (10 μM). B, none of the compounds tested stimulated an agonist response. A glutamate CRC is included as a positive control (●). C, all compounds potentiated the response to a suboptimal (EC20) concentration of glutamate in a concentration-dependent manner. Data represent the mean ± S.E.M. of three to four independent experiments performed in duplicate.
To increase confidence that the difference between responses to the mGlu5 PAMs in the two cell lines is related to differences in receptor expression, we engineered a third HEK293 cell line in which human mGlu5 expression is under control of a tetracycline-inducible promoter. Cells were treated with vehicle or increasing concentrations of tetracycline (1–10 ng/ml) to induce mGlu5 expression. Radioligand binding studies revealed that cells treated with vehicle had low expression of mGlu5 and that increasing concentrations of tetracycline induced corresponding increases in mGlu5 density (Supplemental Fig. 3). VU0360172 is the most potent compound with both agonist and PAM activity and, therefore, was chosen for these studies. As in the previous studies with the lower expression cell line, VU0360172 induced robust potentiation of the response to glutamate in this inducible line when no tetracycline was added and mGlu5 expression was low. However, VU0360172 had no agonist activity in cells in which mGlu5 expression was not induced (Supplemental Fig. 3). With increasing concentrations of tetracycline and corresponding increases in receptor expression, there was a shift from pure PAM activity to agonist activity coupled with PAM activity for VU0360172 (Supplemental Fig. 3). This provides strong evidence that increased expression of mGlu5 can confer ago-PAM activity that is not present in cells expressing lower levels of the receptor. Of importance, the degree of agonism was not as pronounced in this cell line as was observed with the high expression rat mGlu5 cell line (Fig. 2). One possible explanation for the less robust agonism is species differences between rat and human. Because our further studies rely on use of rodent tissue, we focused on use of the rat lower expression cell line.
We further evaluated the ability of the mGlu5 compounds to shift the glutamate concentration-response curve. Incubation of the low expression rat mGlu5 cell line with increasing fixed concentrations of mGlu5 modulators (30 nM–30 μM) followed by a concentration-response curve of glutamate (10 nM–100 μM) resulted in a progressive leftward shifts of the glutamate concentration-response curve for each of the compounds with maximal fold shift values of 6.8-fold at 3 μM VU0360172, 1.9-fold at 30 μM VU0361747, 5.4-fold at 10 μM VU0092273, and 4-fold at 10 μM VU0240382 (n = 3–8) (Fig. 4). In this case, the lower fold shift value for VU0361747 compared with that for the other three compounds corresponds with the lower potency of this compound compared with that of the other mGlu5 compounds studied. Slightly higher fold shift values were observed for all of the compounds with the high expression mGlu5 cell line (VU0360172, 8.7-fold at 100 nM; VU0092273, 9.1-fold at 300 nM; VU0361747, 8.4-fold at 10 μM; and VU0240382, 10.4-fold at 10 μM; n = 3–5) (Supplemental Fig. 4). It was necessary to run the fold shift experiments with VU0360172 and VU0092272 at lower concentrations to avoid the complications of agonist activity when the response in the high mGlu5 expression cell line was assessed. The difference in fold shift values could be a result of differences in coupling efficiency or signaling between the two cell lines. The potential for differences in coupling efficiency or signaling between the cell lines could also explain why in the high expression mGlu5 cell line the compounds elevate the maximum response to glutamate above 100% (Fig. 2D) but are unable to do so in the lower expression cell lines (Fig. 3C).

mGlu5 allosteric modulators induce a leftward shift in the glutamate concentration-response curve in a low expression mGlu5 cell line. Progressive fold shift values were determined by treating cells with increasing fixed concentrations of PAMs followed by the addition of a glutamate CRC. The calcium response was measured and normalized to a maximally effective concentration of glutamate (10 μM). A. VU0360172. B, VU0361747. C, VU0092273. D, VU0240382. Data represent the mean ± S.E.M. of three to eight independent experiments performed in duplicate.
Ago-PAM Activity Is Not Observed in Cortical Astrocytes.
The studies of the effects of mGlu5 PAMs and ago-PAMs in HEK293 cells raise the possibility that ago-PAM activity for these compounds may be a function of overexpression of mGlu5 or other factors that could have an impact on responses in recombinant systems. The availability of new mGlu5 PAMs that are optimized for robust agonist activity provides tools to allow us to determine whether this translates into agonist activity in native systems. As a first method to evaluate the effects of these compounds in a native system, we determined the effects of these pure PAMs and ago-PAMs in rat cortical astrocytes, where mGlu5-mediated responses have been extensively characterized (Peavy and Conn, 1998; Zhang et al., 2005; Chen et al., 2008). Radioligand binding studies revealed that the level of mGlu5 expression in rat cortical astrocytes was similar to that of the low expression mGlu5 HEK cell line (mGlu5 Bmax = 3.4 ± 0.3 fmol/105 cells). Of interest, responses to the mGlu5 PAMs in astrocytes were virtually identical to those observed in the HEK293 cells expressing lower levels of mGlu5 (Fig. 5). Thus, none of the compounds tested induced an mGlu5 agonist response when added alone (Fig. 5A). However, each of the mGlu5 PAMs induced a concentration-dependent potentiation of the response to an EC20 concentration of glutamate with potencies similar to those observed in HEK293 cells (EC50 VU0360172, 80 ± 6 nM; VU0092273, 168 ± 12 nM; VU0240382, 215 ± 48 nM; and VU0361747, 504 ± 98 nM; mean ± S.E.M., n = 3) (Fig. 5B).

mGlu5 allosteric modulators stimulate a functional calcium response in rat cortical astrocytes that is similar to the response observed in the low expression mGlu5 cell line. A and B, to assess the potency of mGlu5 PAMs in rat cortical astrocytes, astrocytes were treated with increasing concentrations of mGlu5 compound before the addition of an EC20 concentration of glutamate. Calcium flux was measured and normalized to a concentration of glutamate that stimulated a maximal response (10 μM). A, an agonist response was not observed with any of the mGlu5 PAMs tested. B, all compounds were able to potentiate the response to an EC20 concentration of glutamate in a concentration-dependent manner. Data represent the mean ± S.E.M. of three independent experiments performed in duplicate.
mGlu5 PAMs Do Not Exhibit Probe Dependence for DHPG.
Studies in astrocytes suggest that members of this class of biaryl acetylene mGlu5 PAMs do not have agonist activity in at least one native system. To further assess activity of these on CNS function, it will be important to evaluate their effects in brain slice preparations. However, it is not practical to use glutamate as an mGlu5 agonist in brain slice electrophysiology studies because it has multiple targets: mGlu receptors, ionotropic glutamate receptors, and glutamate transporters. For studies of mGlu5 responses in brain slices, we routinely use DHPG, a group I orthosteric agonist that selectively activates mGlu1 and mGlu5. However, before advancing to studies with DHPG in brain slices, it is important to determine whether these compounds induce potentiation of responses to DHPG similar as that to glutamate. Previous studies have shown that allosteric modulators can display strong probe dependence and potentiate responses to some but not all orthosteric agonists (Keov et al., 2011). Thus, we used calcium fluorescence assays to determine whether there is a probe-dependent effect of the ability of the compounds to potentiate DHPG compared with glutamate in the high expression mGlu5 cell line. In all cases, the mGlu5 PAMs potentiated the EC20 response to DHPG in a manner similar to that to glutamate (EC50 VU0360172, 6.6 ± 2.7 nM; VU0092273, 34 ± 14 nM; VU0240382, 42 ± 5 nM; VU0361747, 124 ± 16 nM; mean ± S.E.M., n = 4) (Supplemental Fig. 5). These results support the use of DHPG, in place of glutamate, in electrophysiology experiments to study the potentiation of the mGlu5 response by these allosteric modulators.
mGlu5 PAMs Potentiate DHPG-Induced LTD at the Hippocampal SC-CA1 Synapse.
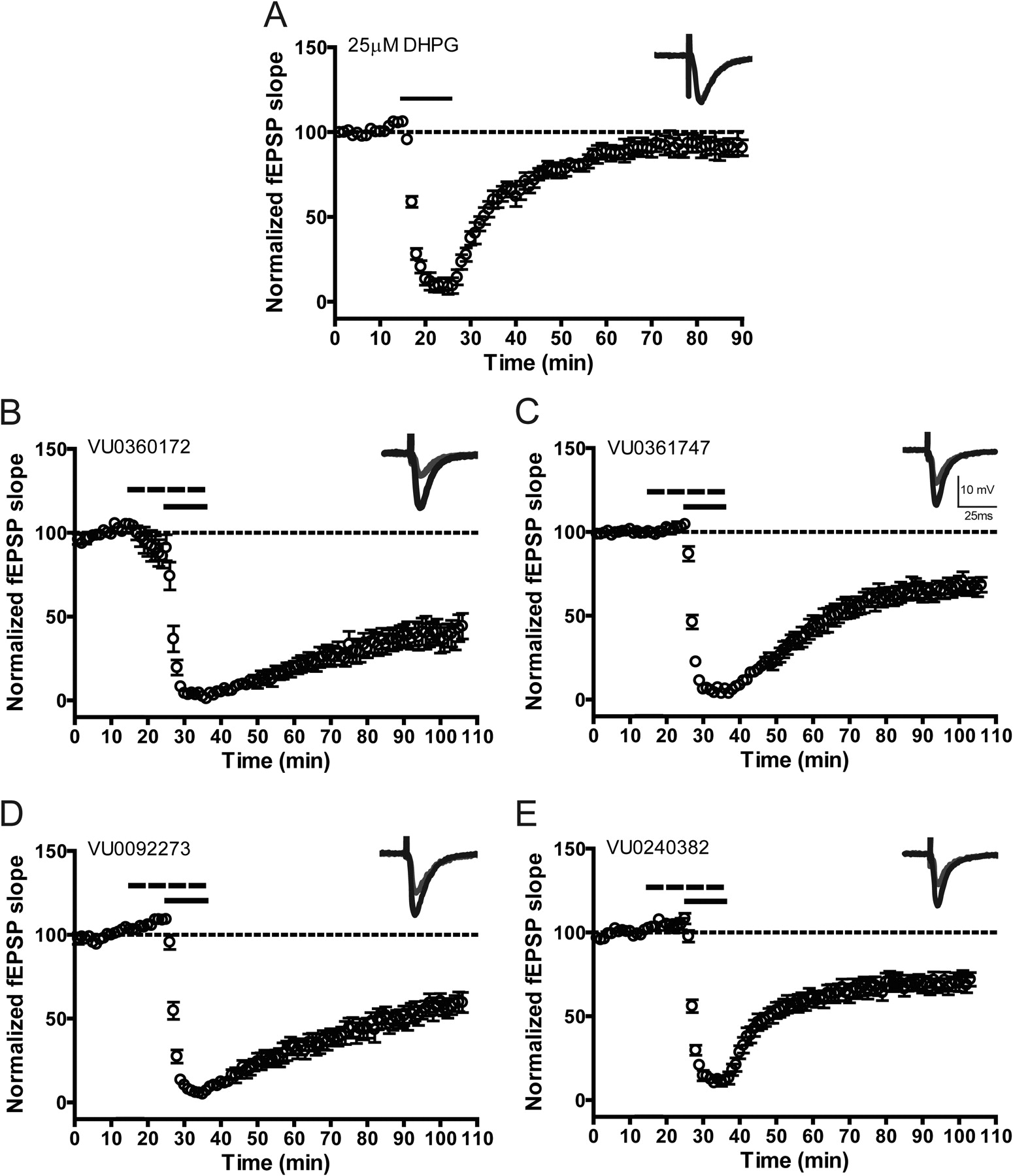
One of the most well characterized responses to mGlu5 activation is induction of a form of synaptic plasticity termed LTD at the SC-CA1 synapse in the hippocampal formation (Huber et al., 2001; Malenka and Bear, 2004; Ayala et al., 2009). Orthosteric mGlu5 agonists induce profound LTD at this synapse, and this can be potentiated by previously reported mGlu5 PAMs (Ayala et al., 2009). If the novel ago-PAMs used in this study have agonist activity at the SC-CA1 synapse, they will be expected to induce LTD when added alone. In contrast, if these compounds act as pure PAMs, they should potentiate responses to DHPG but would not be expected to induce LTD in the absence of agonist. To assess this, extracellular fEPSPs were recorded from the dendritic layer of CA1 after stimulation of the SC-CA1 synapse. In agreement with our previous studies (Ayala et al., 2009), we found that 75 μM DHPG induced robust depression of synaptic responses that persisted at least 55 min after washout (Fig. 6A). LTD was defined as the level of depression 55 min after washout of DHPG (55.3 ± 4.2% of baseline). In contrast to DHPG, 3 μM VU0360172 did not induce a LTD response when added alone (Fig. 6B), with a mean fEPSP slope of 97.1 ± 4.3% of baseline 55 min after compound washout. Likewise, treatment of slices with 10 μM VU0360172 alone did not result in the induction of LTD (fEPSP slope = 91.6 ± 5.1% of baseline), and there was only a small transient decrease in fEPSP slope in two of the seven slices assessed (Supplemental Fig. 6A).
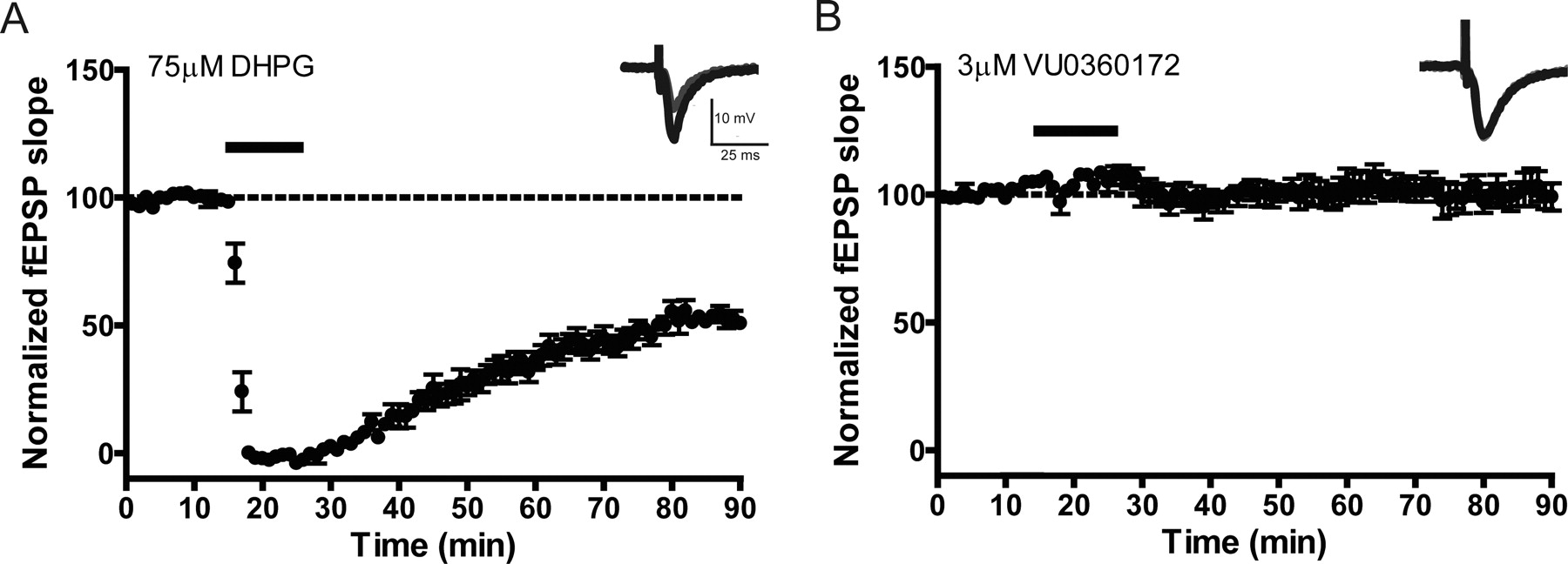
Long-term depression is induced by DHPG but not by VU0360172 alone, at the Schaffer collateral-CA1 synapse in the hippocampus. fEPSPs in which a response of 40 to 60% of the maximal response was used to assess the effect of bath administration of DHPG or VU0360172 at the SC-CA1 synapse in rat hippocampal slices were measured. fEPSP slopes were quantified as described under Materials and Methods. Insets are sample fEPSPs traces measured predrug (black) and 55 min after drug washout (gray). A, bath application of 75 μM DHPG (solid line) for 10 min resulted in induction of LTD measured 55 min after dug washout (n = 6). B, treatment of slices with 3 μM VU0360172 (solid line) had no effect on fEPSPs (n = 7). Error bars represent S.E.M.
We next evaluated the effects of each of the mGlu5 PAMs on fEPSP slope both alone and in the presence of a low concentration of DHPG that was below the concentration needed to elicit LTD. Consistent with our previous report (Ayala et al., 2009), 25 μM DHPG induced acute depression of the fEPSP slope but did not induce LTD measured 55 min after washout (fEPSP slope = 95.7 ± 4.8% of baseline) (Fig. 7A). On the basis of these findings, all subsequent potentiation experiments were conducted with 25 μM DHPG. Each of the mGlu5 PAMs was applied for 10 min followed by coapplication of the PAM and 25 μM DHPG for 10 min. In each case, a 10 μM concentration of the mGlu5 PAM alone (VU0361747, VU0092273, or VU0240382) failed to significantly reduce the fEPSP slope during the 10-min incubation before DHPG. This is consistent with the results with VU0360172 and suggests that these compounds do not have intrinsic agonist activity in inducing LTD. However, each mGlu5 PAM induced a significant (p < 0.001) enhancement of LTD induced by 25 μM DHPG measured 55 min after washout of compound (mean ± S.E.M.): VU0360172, 39.2 ± 6.2% of baseline, n = 7 (Fig. 7B); VU0361747, 63.1 ± 5.0% of baseline, n = 6 (Fig. 7C); VU0092273, 51.6 ± 5.8% of baseline, n = 7 (Fig. 7D); VU0240382, 68.6 ± 4.7% of baseline, n = 8 (Fig. 7E). The mGlu5 antagonist MTEP (10 μM) blocked induction of LTD when 3 μM VU0360172 (97.3 ± 6.0% of baseline) or 10 μM VU0360172 (80.9 ± 3.2% of baseline) was administered in combination with 25 μM DHPG assessed 55 min after washout of the compound (Supplemental Fig. 6B). These results confirm the role of mGlu5 in mediating this response.

mGlu5 allosteric modulators potentiate DHPG-induced long-term depression at the Schaffer collateral-CA1 synapse in hippocampus. fEPSPs were measured in rat hippocampus at the SC-CA1 synapse with a stimulus intensity that produced 40 to 60% of the maximum fEPSP response, determined at the start of each individual experiment. fEPSP slopes were quantified as described under Materials and Methods. Insets are sample fEPSPs traces measured predrug (black) and 55 min after drug washout (gray). A, application of 25 μM DHPG (solid line) for 10 min resulted in a slight decrease in fEPSP slope measured 55 min after drug washout (n = 6). B to E, application of mGlu5 compound to the slice for 20 min (dashed line), first alone and then in combination with 25 μM DHPG (solid line; 10 min), resulted in significant enhancement of DHPG-induced LTD (VU0360172, n = 7; VU0361747, n = 6; VU0092273, n = 7; VU0240382, n = 8) but has no effect on acute depression. Error bars represent S.E.M.
VU0360172 Has No Agonist Activity at mGlu5 in Multiple Brain Regions Assessed Using Slice Electrophysiology.
VU0360172 has the most potent agonist activity of the compounds tested in the high mGlu5 expression cell line. Therefore, we further evaluated this mGlu5 PAM to determine whether it has agonist activity for inducing other known mGlu5-mediated responses. mGlu5 is localized postsynaptically on STN neurons and activation of mGlu5 in these neurons leads to depolarization (Awad et al., 2000). Previous work has shown that mGlu5 PAMs N-(1,3-diphenyl-1H-pyrazol-5-yl)-4-nitrobenzamide (VU29), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide, and N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide potentiate membrane depolarization induced by DHPG in STN neurons (O'Brien et al., 2004; Rodriguez et al., 2005; Chen et al., 2007). Consistent with our previous reports, 100 μM DHPG resulted in a robust depolarization of STN neurons (12.1 ± 2.1 mV; mean ± S.E.M., n = 6) (Supplemental Fig. 7). In contrast, bath application of 10 μM VU0360172 had no effect on membrane voltage (0.6 ± 0.7 mV, mean ± S.E.M., n = 5). In addition, 10 μM VU0360172 had no effect on spontaneous firing of STN neurons, whereas DHPG induced a robust increase in spontaneous firing in STN neurons (data not shown). Further experiments were undertaken in medium spiny neurons in striatum, which have been shown to express higher levels of mGlu5 than STN neurons (Testa et al., 1995; Kerner et al., 1997; Pisani et al., 2001). Once again, 10 μM VU0360172 did not mimic the established effects of DHPG in these cells (data not shown). Taken together these results suggest that VU0360172 does not have intrinsic mGlu5 agonist activity in any of the native systems studied.
VU0360172, VU0361747, and VU0092273 Significantly Reduced Amphetamine-Induced Hyperlocomotion.
The primary impetus for discovery and characterization of mGlu5 PAMs has been their potential use as a treatment for schizophrenia. A well characterized in vivo behavioral assay that has been used to evaluate the potential antipsychotic efficacy of mGlu5 PAMs is assessing their ability to reverse amphetamine-induced hyperlocomotor activity in rodents (Kinney et al., 2005; Liu et al., 2008; Rodriguez et al., 2010). Of interest, all mGlu5 PAMs that have previously been evaluated for activity in animal models that predict antipsychotic activity possessed ago-PAM activity. Thus, it is not clear whether agonist activity as measured in cell lines is an important property to confer in vivo efficacy in animal models. Therefore, we determined the effects of the mGlu5 ago-PAMs, VU0360172 and VU0092273, and the pure PAM, VU0361747, on amphetamine-induced hyperlocomotor activity. Consistent with the findings of Rodriguez et al. (2010), administration of VU0360172 significantly (p < 0.0001) reversed amphetamine-induced hyperlocomotion when analyzed from the time of amphetamine delivery to the end of testing period (60–120 min). Thus, the group receiving 56.6 mg/kg i.p. VU0360172 before amphetamine administration produced significantly fewer ambulations (p < 0.0001) than the group receiving vehicle and amphetamine. This result represents a 56% reduction in the hyperlocomotor response (Fig. 8A). Furthermore, VU0092273, another ago-PAM in in vitro assays, also significantly (p < 0.0001) reduced the increase in locomotor activity caused by subcutaneous administration of amphetamine with a maximum reversal of 60% (Fig. 8C). Finally, the pure mGlu5 PAM, VU0361747 (56.6 mg/kg), significantly reversed amphetamine-induced hyperlocomotion in a manner similar to that observed with the compounds that possess ago-PAM activity in HEK cells expressing high levels of mGlu5. Ambulations in rats receiving VU0361747 were reduced by 53% (Fig. 8B). These data suggest that compounds optimized as pure PAMs have similar activity in this animal model compared with that of compounds optimized to have robust ago-PAM activity in overexpressing cell lines.
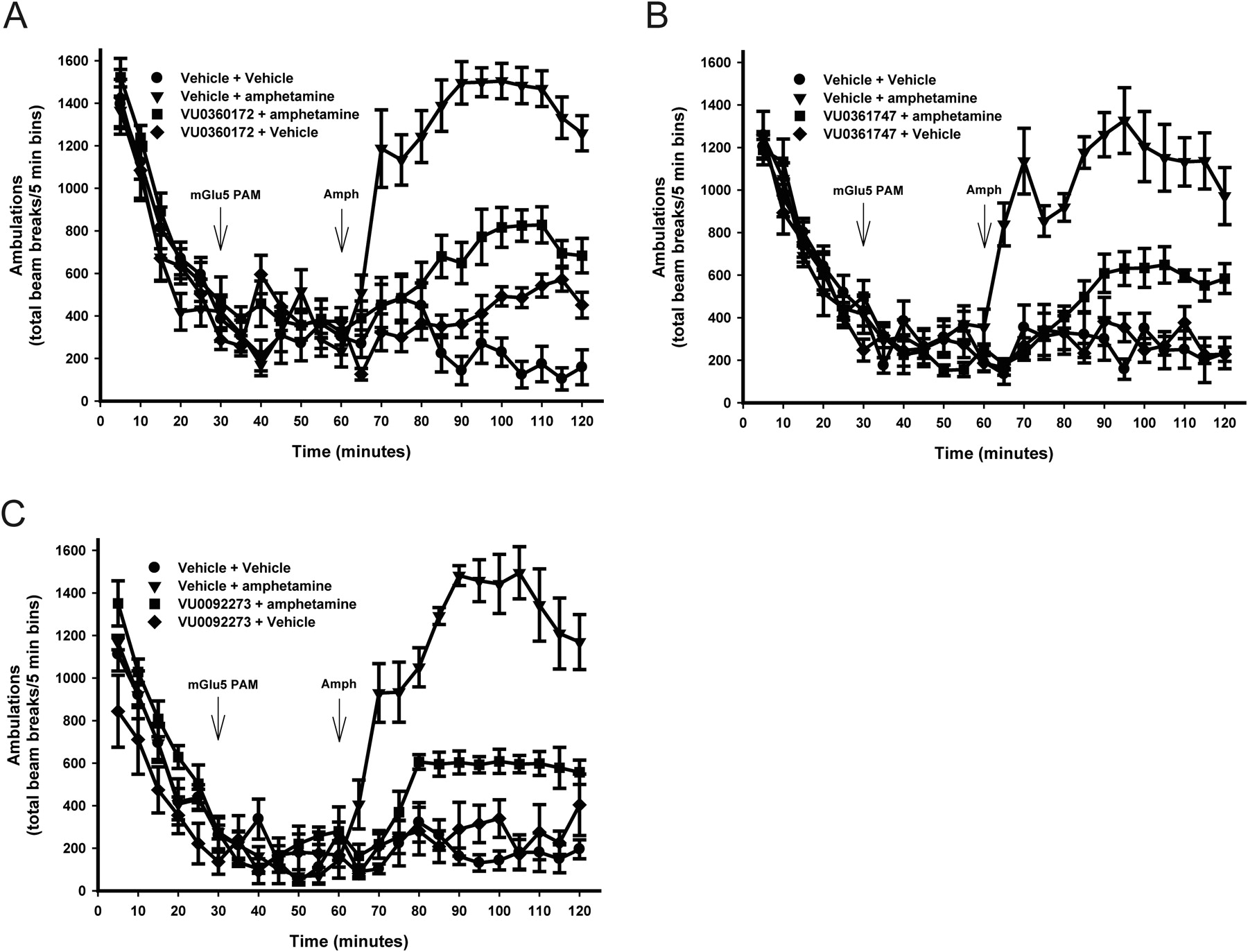
mGlu5 PAMs reverse amphetamine (Amph)-induced hyperlocomotion. Rats were placed in the open-field chambers for a 30-min habituation interval, followed by a pretreatment with a 10% Tween 80 vehicle or a 56.6 mg/kg dose of test compound intraperitoneally for an additional 30 min. Rats then received an injection of saline or 1 mg/kg amphetamine subcutaneously, and locomotor activity was measured for an additional 60 min. Statistical analyses were performed using JMP8 (SAS Institute Inc., Cary, NC). VU0360172 (A), VU0361747 (B), and VU0092273 (C) significantly reduced amphetamine-induced hyperlocomotion. Data are reported as mean ± S.E.M. of the total number of beam breaks per 5-min intervals (n = 5–12/dose).
Discussion
Preclinical studies suggest that selective activation of mGlu receptors may be beneficial for the treatment of various disorders of the CNS. The current focus is on identifying compounds that act at allosteric sites on these receptors, which are less conserved across subtypes, to achieve a greater degree of subtype selectivity. In a previous study, we reported that allosteric modulators of mGlu5 can display a range of activities from positive allosteric modulator to negative allosteric modulator to neutral cooperativity with slight structural modifications (O'Brien et al., 2004; Rodriguez et al., 2005, 2010). In some cases, individual scaffolds can be strongly biased toward dominant positive or negative allosteric modulator activity; however, additional chemical exploration has shown that for some series, it is possible to identify novel opposing molecular switches that can completely alter the ligand's pharmacological function (Sharma et al., 2009; Schann et al., 2010). We recently reported results of an mGlu5 high-throughput screen resulting in the identification of VU0092273 (Rodriguez et al., 2010) and related monocyclic and bicyclic biaryl acetylene compounds, some of which are the most potent mGlu5 PAMs to date. In addition, these agents can have different profiles in different cell populations, such as pure PAM or ago-PAM activity. This intrinsic agonist or ago-PAM activity has been observed for a number of mGlu5 PAMs across multiple laboratories, but the functional relevance of the agonist activity of mGlu5 PAMs has not been clear.
In the present studies, we took advantage of novel mGlu5 PAMs that were systematically optimized and evaluated to favor strong agonist activity and closely related compounds that did not display robust agonist activity in the primary cell lines (high expression) that have been used for discovery of novel mGlu5 PAMs. Using these compounds and new cell lines engineered to have lower levels of mGlu5 expression, we found that ago-PAM activity is only observed in cell lines expressing higher levels of mGlu5. Furthermore, we used a separate cell line in which human mGlu5 is expressed under an inducible promoter and found that, consistent with our observations in the high and low expression rat mGlu5 cell lines, the most potent ago-PAM, VU0360172, has agonist activity when mGlu5 expression is induced to high levels but has pure PAM activity when mGlu5 expression is maintained at lower levels. These studies suggest that overexpression of mGlu5 in cell lines may be one factor that contributes to the presence of ago-PAM activity for some allosteric modulators. However, whereas overexpression of mGlu5 may be an important factor in the observation of ago-PAM activity, other factors may also contribute to the ability of some compounds to directly activate the receptor along with some that strictly depend on activation by glutamate.
Although it is impossible to definitively relate expression levels to individual neuronal populations or subcellular compartments in neurons, it is possible that certain locations in the brain may exhibit higher levels of mGlu5 relative to those in other areas. In addition, other factors that may influence the ability of some compounds to act as ago-PAMs and others as pure PAMs could exist in native systems. Thus, it will be important to evaluate whether the agonist activity observed in vitro under conditions of high receptor expression may be relevant and may be critical for in vivo activity. Further experiments revealed that optimized mGlu5 ago-PAMs and pure PAMs behave similarly in multiple native systems, including studies in rat cortical astrocytes, measures of effects on hippocampal LTD, and depolarization of STN neurons. In these native systems, all compounds behaved as mGlu5 PAMs, but none showed evidence of direct mGlu5 agonist activity. Finally, one of the most important findings of the present studies is that a compound that behaved as a pure PAM in mGlu5 cell lines, VU0361747, exhibited similar efficacy in at least one animal model compared with other compounds that have robust agonist activity in the cell line assays (VU0360172 and VU0092273). Taken together, these data suggest that the agonist activity of mGlu5 PAMs may be influenced by high expression in recombinant cell lines and does not necessarily predict agonist activity in native systems. Furthermore, agonist activity in expression systems may not be an important factor in determining in vivo efficacy.
Despite the finding that each of these compounds behaves in a similar manner in the native systems studied here, it would not be appropriate to conclude that allosteric agonist activity will not be observed in any native system. It is possible that there are specific neuronal populations or subcellular compartments where mGlu5 levels are higher than those studied here or where other factors provide a context by which some PAMs can display ago-PAM activity in native systems. In addition, it is important to remain open to the possibility that compounds with agonist activity in overexpressing cell lines will differentiate from pure PAMs in other behavioral assays. Previous studies have shown that mGlu5 PAMs have a number of in vivo effects in multiple animal models that are relevant for schizophrenia and cognition-enhancing activity. It is possible that these compounds will have differential responses when assessed across a broad range of physiological responses and behavioral models. However, the studies presented here clearly suggest that the predominant effect of mGlu5 PAMs in native systems is to potentiate responses to glutamate rather than directly activating the receptor. Of importance, these compounds provide new tools that can be used to fully address these important questions across a broader range of measures of mGlu5 function.
An especially interesting component of these studies was the finding that compounds classified as strong ago-PAMs versus pure PAMs did not differentiate in studies of LTD. DHPG-induced LTD at the SC-CA1 synapse in hippocampus has been shown to be mediated primarily by mGlu5 (Huber et al., 2001; Faas et al., 2002; Huang and Hsu, 2006). In addition, mGlu5 plays an important role in the induction of long-term potentiation (LTP) at the same synapse (Lu et al., 1997; Cohen et al., 1998; Ayala et al., 2009). A balance of LTP and LTD is thought to be critical for normal cognitive function, and any manipulation that disrupts this balance could impair cognition. This factor is especially critical in considering development of mGlu5 activators as therapeutic agents because orthosteric mGlu5 agonists induce profound LTD and disrupt this critical balance between LTD and LTP. On the basis of this finding, overactivity of mGlu5 has been postulated to play an important role in impairments in cognitive function in fragile X syndrome, in which signaling of this receptor is altered (Huber et al., 2002; Bear et al., 2004; Lauterborn et al., 2007). Of interest, we previously reported that other mGlu5 PAMs, such as VU29, enhance both LTD and LTP but maintain a strict dependence of both on specific patterns of afferent activity and maintain the balance between these opposing forms of synaptic plasticity (Ayala et al., 2009). This unique profile may be critical for the cognition-enhancing effects of these agents and is likely to depend on the mechanism of these compounds in potentiating but not directly activating mGlu5. However, there has been a concern that compounds that have strong ago-PAM activity in vitro may not provide this important advantage and could induce strong LTD in a manner similar to that of orthosteric agonists. The current finding that compounds classified from in vitro studies as strong ago-PAMs had effects on LTD similar to those of compounds classified as pure PAMs was encouraging and suggests that this difference in activity profile in cell lines may not predict adverse effects on cognitive function. In future studies, it will be important to evaluate these mGlu5 PAMs in animal models of learning and memory to determine whether they have similar effects in enhancing hippocampal-dependent cognitive function.
In conclusion, the current studies provide initial evidence that the mGlu5 ago-PAM activity that is often observed in cell-based assays may not reflect a clear distinction between pure PAMs and ago-PAMs that is relevant to the activity of these compounds in native systems or to their in vivo efficacy. The apparent discrepancy between in vitro and in vivo, in terms of the mode of action (PAM versus ago-PAM), may be due in part to the artificial nature of cell expression systems in which overexpression of the receptor may lead to differences in interactions between receptors, receptor coupling, and the signaling pathways activated. It is important to mention that this does not seem to be a strict example of the well known phenomenon of receptor reserve in that the orthosteric agonist glutamate exhibited similar potencies in cell lines expressing different mGlu5 densities. If the high expression cell lines had high receptor reserve according to the classic view, the glutamate CRC would be expected to be shifted to the left with higher mGlu5 expression. However, this could represent an analogous phenomenon that influences the ability to detect agonist responses of mGlu5 PAMs. In future studies, it will be important to gain a further understanding of the mechanistic underpinnings of these differences in mGlu5 PAM activity and to also further evaluate the relevance of these differences in other native systems.
Authorship Contributions
Participated in research design: Noetzel, Rook, Days, Rodriguez, Lavreysen, Niswender, Xiang, Weaver, Daniels, Jones, Lindsley, and Conn.
Conducted experiments: Noetzel, Rook, Vinson, and Cho.
Contributed new reagents or analytic tools: Zhou and Stauffer.
Performed data analysis: Noetzel, Rook, and Vinson.
Wrote or contributed to the writing of the manuscript: Noetzel, Rook, Stauffer, and Conn.
Acknowledgments
We thank Ryan Morrison and Kiran Gogi for their technical assistance.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Institutes of Health National Institute of Mental Health [Grant 2R01-MH062646-12]; National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants 2R01-NS031373-16A2, F32-NS071746]; and National Institutes of Health National Institute of Mental Health Molecular Libraries Probe Production Centers Network [Grants 5 u54 MH84659-03, 5 u54 MH84659-03S1].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- mGlu
- metabotropic glutamate receptor
- CNS
- central nervous system
- NMDA
- N-methyl-d-aspartate
- PAM
- positive allosteric modulator
- ago-PAM
- allosteric agonist coupled with PAM activity
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- DHPG
- dihydroxyphenylglycine
- VU0360172
- N-cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide
- VU0361747
- (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(4-hydroxypiperidin-1-yl)methanone
- VU0092273
- (4-hydroxypiperidin-1-yl)(4-phenylethynyl)phenyl)methanone
- VU0240382
- 6-(2-phenylethynyl)-1,2,3,4-tetrahydroisoquinolin-1-one
- MTEP
- 3-((2-methyl-4-thiazolyl)ethynyl)pyridine
- HEK
- human embryonic kidney
- GIRK
- G protein-coupled inwardly rectifying potassium channels
- DMSO
- dimethyl sulfoxide
- HBSS
- Hanks' balanced salt solution
- CRC
- concentration-response curve
- AGM
- assay growth media
- l-AP4
- l-(+)-2-amino-4-phosphonobutyric acid
- aCSF
- artificial cerebrospinal fluid
- fEPSP
- field excitatory postsynaptic potential
- LTD
- long-term depression
- STN
- subthalamic nucleus
- HPLC
- high-performance liquid chromatography
- SC-CA1
- Schaffer collateral-CA1
- VU29
- N-(1,3-diphenyl-1H-pyrazol-5-yl)-4-nitrobenzamide
- LTP
- long-term potentiation.

- Received August 8, 2011.
- Accepted October 20, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}